10-K: Annual report pursuant to Section 13 and 15(d)
Published on March 28, 2016
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
|
x |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
FOR THE FISCAL YEAR ENDED DECEMBER 31, 2015
|
o |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
FOR THE TRANSITION PERIOD FROM TO .
COMMISSION FILE NUMBER: 001-37348
Corbus Pharmaceuticals Holdings, Inc.
(Exact name of registrant as specified in its charter)
|
Delaware |
|
46-4348039 |
|
(State or other jurisdiction of incorporation or organization) |
|
(I.R.S. Employer Identification No.) |
|
|
|
|
|
100 River Ridge Drive Norwood, Massachusetts |
|
02062 |
|
(Address of principal executive offices) |
|
(Zip Code) |
(617) 963-0100
Registrant’s telephone number, including area code:
Securities registered pursuant to Section 12(b) of the Act: None
Securities registered pursuant to Section 12(g) of the Act:
|
Title of each class |
|
Common Stock, par value $0.0001 per share |
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
|
Large accelerated filer |
o |
|
Accelerated filer |
o |
|
|
|
|
|
|
|
Non-accelerated filer |
o |
(Do not check if a smaller reporting company) |
Smaller reporting company |
x |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No x
As of June 30, 2015, the last business day of the registrant’s most recently completed second fiscal quarter, the aggregate market value of the common stock held by non-affiliates of the registrant was approximately $72,826,849, based on the closing price of the registrant’s common stock on June 30, 2015.
As of March 24, 2016, the number of shares outstanding of the registrant’s common stock, $0.0001 par value per share, was 37,605,827.
Documents incorporated by reference
Portions of the registrant’s proxy statement for the 2016 annual meeting of stockholders to be filed pursuant to Regulation 14A within 120 days after the registrant’s fiscal year ended December 31, 2015, are incorporated by reference in Part III of this Form 10-K.
CORBUS PHARMACEUTICALS HOLDINGS, INC.
ANNUAL REPORT ON FORM 10-K
FOR THE YEAR ENDED DECEMBER 31, 2015
TABLE OF CONTENTS
|
ITEM |
|
|
|
Page |
|
|
|
PART I |
|
|
|
1. |
|
|
2 |
|
|
1A. |
|
|
23 |
|
|
1B. |
|
|
43 |
|
|
2. |
|
|
43 |
|
|
3. |
|
|
43 |
|
|
4. |
|
|
43 |
|
|
|
|
|
|
|
|
|
|
PART II |
|
|
|
5. |
|
|
44 |
|
|
6. |
|
|
44 |
|
|
7. |
|
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
|
45 |
|
7A. |
|
|
50 |
|
|
8. |
|
|
50 |
|
|
9. |
|
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
|
50 |
|
9A. |
|
|
50 |
|
|
9B. |
|
|
51 |
|
|
|
|
|
|
|
|
|
|
PART III |
|
|
|
10. |
|
|
52 |
|
|
11. |
|
|
52 |
|
|
12. |
|
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
|
52 |
|
13. |
|
Certain Relationships and Related Transactions, and Director Independence |
|
52 |
|
14. |
|
|
52 |
|
|
|
|
|
|
|
|
|
|
PART IV |
|
|
|
15. |
|
|
53 |
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This report on Form 10-K contains forward-looking statements made pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995 under Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. Forward-looking statements include statements with respect to our beliefs, plans, objectives, goals, expectations, anticipations, assumptions, estimates, intentions and future performance, and involve known and unknown risks, uncertainties and other factors, which may be beyond our control, and which may cause our actual results, performance or achievements to be materially different from future results, performance or achievements expressed or implied by such forward-looking statements. All statements other than statements of historical fact are statements that could be forward-looking statements. You can identify these forward-looking statements through our use of words such as “may,” “can,” “anticipate,” “assume,” “should,” “indicate,” “would,” “believe,” “contemplate,” “expect,” “seek,” “estimate,” “continue,” “plan,” “point to,” “project,” “predict,” “could,” “intend,” “target,” “potential” and other similar words and expressions of the future.
There are a number of important factors that could cause the actual results to differ materially from those expressed in any forward-looking statement made by us. These factors include, but are not limited to:
|
|
· |
our lack of operating history; |
|
|
· |
our current and future capital requirements and our ability to satisfy our capital needs; |
|
|
· |
our ability to complete required clinical trials of our product and obtain approval from the FDA or other regulatory agents in different jurisdictions; |
|
|
· |
our ability to maintain or protect the validity of our patents and other intellectual property; |
|
|
· |
our ability to retain key executive members; |
|
|
· |
our ability to internally develop new inventions and intellectual property; |
|
|
· |
interpretations of current laws and the passages of future laws; |
|
|
· |
acceptance of our business model by investors; |
|
|
· |
the accuracy of our estimates regarding expenses and capital requirements; and |
|
|
· |
our ability to adequately support growth. |
The foregoing does not represent an exhaustive list of matters that may be covered by the forward-looking statements contained herein or risk factors that we are faced with that may cause our actual results to differ from those anticipate in our forward-looking statements. Please see “Risk Factors” for additional risks which could adversely impact our business and financial performance.
All forward-looking statements are expressly qualified in their entirety by this cautionary notice. You are cautioned not to place undue reliance on any forward-looking statements, which speak only as of the date of this report or the date of the document incorporated by reference into this report. We have no obligation, and expressly disclaim any obligation, to update, revise or correct any of the forward-looking statements, whether as a result of new information, future events or otherwise. We have expressed our expectations, beliefs and projections in good faith and we believe they have a reasonable basis. However, we cannot assure you that our expectations, beliefs or projections will result or be achieved or accomplished.
1
Overview
This report and the information incorporated herein by reference contain references to trademarks, service marks and trade names owned by us or other companies. Solely for convenience, trademarks, service marks and trade names referred to in this report and the information incorporated herein, including logos, artwork, and other visual displays, may appear without the ® or ™ symbols, but such references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights or the rights of the applicable licensor to these trademarks, service marks and trade names. We do not intend our use or display of other companies’ trade names, service marks or trademarks to imply a relationship with, or endorsement or sponsorship of us by, any other companies. Other trademarks, trade names and service marks appearing in this report are the property of their respective owners.
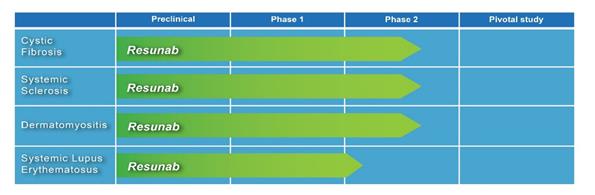
We are a clinical stage pharmaceutical company, focused on the development and commercialization of novel therapeutics to treat rare or uncommon chronic and serious inflammatory and fibrotic diseases with clear unmet medical needs. Our product Resunab is a novel synthetic oral endocannabinoid-mimetic drug that is designed to resolve chronic inflammation and halt fibrotic processes without causing immunosuppression. In preclinical models, Resunab also demonstrated an ability to promote bacterial clearance by the immune system. Resunab is currently being evaluated in three separate Phase 2 studies for the treatment of cystic fibrosis, or CF, systemic sclerosis and skin-predominant dermatomyositis. Top-line data from the cystic fibrosis and systemic sclerosis studies are anticipated by the end of 2016 and from the dermatomyositis study in the first quarter of 2017. The United States Food and Drug Administration (the “FDA”) has granted Resunab Orphan Drug Designation as well as Fast Track Status for both cystic fibrosis and systemic sclerosis. A fourth Phase 2 study of Resunab in systemic lupus erythematosus, or SLE, is planned to start in the first quarter of 2017.
Since our inception, we have devoted substantially all of our efforts to business planning, research and development, recruiting management and technical staff, acquiring operating assets and raising capital. Our research and development activities have included conducting pre-clinical studies, developing manufacturing methods and manufacturing of our lead drug Resunab for clinical trials and conducting clinical studies in patients.
Resunab is a synthetic, rationally-designed oral small molecule drug that selectively binds to the cannabinoid receptor type 2, or CB2, which is found on activated immune cells, fibroblasts and muscle cells. Resunab stimulates the production of Specialized Pro-Resolving Lipid Mediators, or SPMs, which act to resolve inflammation, clear bacteria and halt fibrosis by activating endogenous pathways. These endogenous resolution pathways are normally activated in healthy individuals during the course of normal immune responses but are dysfunctional in chronic inflammatory and fibrotic diseases. Through its’ activation of the CB2 receptor, Resunab is designed to move innate immune responses from the activation phase through completion of the resolution phase. The CB2 receptor plays an endogenous role in modulating and resolving inflammation by, in effect, turning heightened inflammation “off” and restoring homeostasis.
A key aspect of the body’s innate immune response in the activation phase is the recruitment of inflammatory cells to the site of tissue infection/injury whereupon these cells act to destroy the infection and/or repair tissue damage. The activation phase of the innate immune response prepares the body for more effective clearance of the infectious organism and repair of damaged tissue. The next phase in a normal innate immune response is the resolution phase, during which the nature of the infiltrating immune cells changes from inflammatory to non-inflammatory, the infectious organisms are eliminated as a threat, the tissue then is cleared of residual cellular debris and immune cells, and tissue repair processes stop. In chronic inflammatory and fibrotic diseases, the innate immune responses are “stuck” in the initial activation phase. This failure to progress through the resolution phase causes chronic tissue infiltration with inflammatory cells and chronic activation of healing processes that cause tissue scarring, or fibrosis. The key event that propels an innate immune response from its activation phase to its resolution phase is a “class switch” from production of pro-inflammatory lipid mediators such as prostaglandins and leukotrienes to a family of SPMs (Figure 1) which include lipoxins, resolvins, protectins, and marescins. If an innate immune response persists in the activation phase and does not progress through resolution, chronic inflammation and fibrosis can result, causing organ dysfunction, organ failure, severe morbidity and even death. There are hundreds of inflammatory diseases, many of which are chronic, life-long and incurable.
2
Figure 1. Resunab’s Intended Mechanism of Action
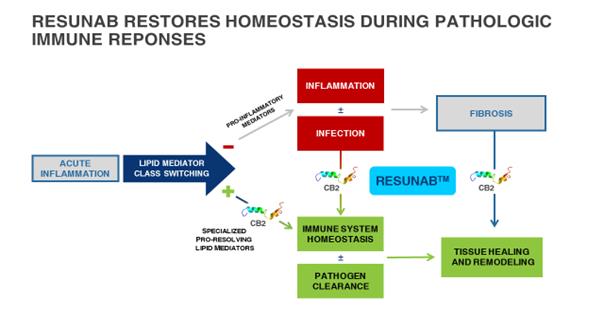
Resunab is designed to restore immune system homeostasis on a daily basis, by using the body’s own physiologic pathways to transition an innate immune response from the activation phase to the resolution phase. If an innate immune response is “stuck” in the activation phase, tissue damage, fibrosis and persistent infection are expected consequences. Endogenous progression of the innate immune response through its resolution phase has been shown to clear inflammation, stop fibrosis, and promote pathogen clearance. Resunab’ s unique mechanism of action is very different than anti-inflammatory drugs which inhibit production or functions of certain pro-inflammatory mediators that initiate or are active during the activation phase. Activation of an innate immune response is necessary to clear infections, so that drugs that interfere with the activation phase carry the risk of immunosuppression and may have other undesirable side effects. In contrast, Resunab is designed to transition an innate immune response from its activation phase to resolution phase. Resunab’s CB2 agonist activity initiates a class switch in bioactive lipid mediators from inflammation-activating mediators to pro-resolving mediators.
Preclinical studies in isolated cells and animal models of disease and clinical pilot studies have shown that Resunab triggers the transition from the activation phase to the resolution phase of innate immune responses. Resunab shifts the balance of lipid mediators to favor production of SPMs and anti-inflammatory lipid mediators (such as prostaglandin J2) versus inflammation-activating lipid mediators (such as leukotriene B4 and prostaglandin E2). The transition out of the activation phase is reflected in preclinical cellular or animal models by reduced tissue edema, swelling, tenderness and pain, reduced production of the chemokine interleukin-8, and reduced production of inflammatory cytokines such as interleukin-6, tumor necrosis factor-α, interleukin-1β, and type 1 interferons. The transition into and through the resolution phase is reflected in preclinical cellular or animal models by increased clearance of bacteria (Pseudomonas aeruginosa from the lung), apoptosis of T cells, reduction of neutrophils and a normalization of the number of macrophages in inflamed tissues. Production of transforming growth factor (TGFβ), myofibroblast accumulation, collagen production, and fibrosis are all reduced in tissue in pre-clinical animal models.
3
The development status of Resunab is summarized below:
Figure 2: Drug developmental pipeline
Market Opportunity in Inflammatory and Fibrotic Diseases
Chronic, serious inflammatory and fibrotic diseases are varied. Some examples of chronic, serious diseases characterized by inflammation with variable degrees of fibrosis include genetic diseases such as cystic fibrosis, nonalcoholic steatohepatitis, or NASH, autoimmune diseases including systemic sclerosis, systemic lupus erythematosus, myositis, rheumatoid arthritis, vasculitis, primary biliary cirrhosis and lung diseases including idiopathic pulmonary fibrosis, bronchiolitis obliterans, and sarcoidosis.
According to Global Business Intelligence Research, the global market for drugs to treat chronic inflammation in 2010 was approximately $58 billion and is expected to grow to approximately $86 billion by 2017. While some chronic inflammatory diseases are very common, for example, about 28.5 million Americans have chronic sinusitis and about 18.7 million Americans have asthma, our initial focus is on chronic, serious inflammatory and fibrotic diseases that are rare or uncommon and have significant unmet medical need. Some of these diseases can be categorized as “orphan diseases” in the U.S., meaning they affect no more than 200,000 patients each. Examples include cystic fibrosis, systemic sclerosis and idiopathic pulmonary fibrosis.
Markets for Resunab
Cystic Fibrosis
Cystic fibrosis is a life-long, progressive, debilitating, and life-threatening autosomal recessive disease. Cystic fibrosis is caused by mutations in the gene Cystic Fibrosis Transmembrane Conductance Regulator or CFTR. The CFTR serves as a central hub to modulate transport, trafficking, and signaling in cells. Given multiple roles of CFTR in cellular activation and homeostasis, mutation of the CFTR give rise to multiple disorders in respiratory, digestive and reproductive organs.
The CFTR mutations lead to defective ion transport, with reduced chloride and bicarbonate secretion and sodium hyper-absorption, followed by water hyper-absorption, by airway epithelia and other cell types. The resultant reduced height of epithelial lining fluid and decreased hydration of mucus results in abnormally thick and sticky mucus, which obstructs the lumen into which the mucus is secreted and reduces mucociliary clearance of bacteria. The dysfunction in ion transport in CF patients is reflected in abnormal sweat chloride levels.
The negative effects caused by CFTR mutations are not restricted to ion channels, but also extend to dysfunction of the innate immune system. The nature of the abnormalities in CF are consistent with inability of innate immune responses to make the transition out of the activation phase and into and through the resolution phase. Bioactive lipid mediators (SPMs) that initiate the transition to resolution phase of innate immune responses have been found to be deficient relative to pro-inflammatory lipid mediators that initiate its activation phase, and this reduction correlates with poor recovery of lung function following an acute pulmonary exacerbation in children. The preponderance of activated neutrophils and pro-inflammatory macrophages in inflamed tissue, reduced neutrophil apoptosis, high levels of neutrophil proteases that reflect persistent neutrophil activation, reduced clearance of neutrophils by macrophages, ineffective clearance of certain bacteria such as P. aeruginosa, and excessive activation of fibrotic pathways all show the inability of individuals with CF to resolve their innate immune responses.
4
An overview of the disease progression in cystic fibrosis is provided in Figure 3.
Figure 3: Factors involved in cystic fibrosis progression
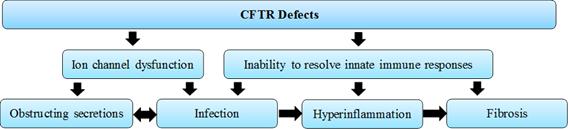
As a result of obstructing secretions, recurrent infections, hyper-inflammation, and activated fibrotic pathways in the lungs, individuals with CF develop bronchiectasis, pulmonary fibrosis, mixed obstructive/restrictive lung disease, and, eventually, respiratory failure. They may also have chronic sinusitis and nasal polyps. The same pathophysiologic events of obstruction, infection, chronic inflammation, and tissue damage/fibrosis occur in the gastrointestinal system, which can lead to bowel obstructions, fat malabsorption, bacterial overgrowth, gut dysmotility, malnutrition, growth retardation, low weight, pancreatic insufficiency, cystic fibrosis-related diabetes, gallstones, and liver failure including cirrhosis. Adult males with cystic fibrosis have degeneration of the ductus deferens and sterility. End-stage organ involvement in cystic fibrosis is sometimes treated with transplantation, especially lung transplantation.
The median current life expectancy of cystic fibrosis patients is about 40 years. According to the Cystic Fibrosis Foundation, 30,000 Americans and a total of 70,000 people in the United States and Europe suffer from cystic fibrosis. The cost of treating cystic fibrosis is high, with an average cost in the U.S. of between $50,000- $74,000 per patient per annum.
Current therapies for cystic fibrosis include mucolytics to breakdown mucus, antibiotics to fight bacterial infection, and, very recently, drugs that act to restore some functionality to the faulty CFTR protein in specific genetic sub-populations of patients, including Kalydeco™ and Orkambi™. Kalydeco™ was approved for treatment of cystic fibrosis in January 2012 and Orkambi was approved in June 2015. With about 2,000 different known mutations in the CFTR gene, Kalydeco™ can only be used by a specific sub-section of the cystic fibrosis population who suffer from a limited number of “gating mutations,” including the G551D mutation. Combined, these currently account for about 5.7% of the total cystic fibrosis patient population worldwide. Orkambi™ is currently only approved for adult patients carrying two copies of the delta508 CFTR mutation.
Of importance, drugs that are sufficient to correct ion channel functions of mutant CFTR protein are not necessarily able to correct the dysfunction of the innate immune system. For example, ivacaftor treatment has not been associated with reduction in sputum neutrophils or neutrophil derived proteases in CF patients. Thus correction of ion channel dysfunction does not necessarily translate into correction of dysfunction of the innate immune system.
All CF patients appear to have dysfunction in resolution of the innate immune system, no matter which CFTR mutations a given patient has. Currently, there is no drug that is used commonly to this basic problem in CF, other than antibiotics to control infection and, indirectly, control inflammation. The use of high dose ibuprofen as an anti-inflammatory treatment is limited to about 3-5% of CF patients since there is a need to monitor levels closely in children and due to side effect risks, primarily gastrointestinal bleeding. The Cystic Fibrosis Foundation Strategic Plan Report, 2014-2018, includes a strategic priority to identify new treatments for CF that allow all patients to better manage the symptoms of their disease and improve their health, with a specific mention of the need for new agents that can resolve inflammation.
We believe that there is general agreement in the CF community that an effective drug that will control hyper-inflammation and help clear infections would address a significant unmet medical need in CF, especially a drug that is orally administered, can be used chronically with other commonly prescribed medications for CF, is not immunosuppressive, and has anti-fibrotic effects.
(Systemic Sclerosis) Scleroderma
Systemic sclerosis is a chronic, systemic autoimmune disease characterized by activation of innate and specific immune systems, an obliterative, proliferative vasculopathy of small blood vessels, and fibrosis of the skin and multiple internal organs. Approximately 90,000 people in the United States and Europe have systemic sclerosis. The disease affects mainly adults (80% of systemic sclerosis patients are women) with mean age of onset about 46 years of age in the United States and the majority of patients between 45-64 years of age.
5
A commonly used system classifies systemic sclerosis patients into those with more wide-spread skin thickening (diffuse cutaneous systemic sclerosis, about 45% of patients) and those with more restricted skin thickening (limited cutaneous systemic sclerosis, about 55% of patients). There is significant overlap in the clinical manifestations for these two groups of systemic sclerosis patients and no known significant differences in disease pathogenesis.
Systemic sclerosis can affect multiple internal organs in the body, including the lungs, heart, kidneys, joints, muscles, esophagus, stomach and intestines. Clinically apparent organ involvement that occurs in more than a third of these patients includes thickened skin, Raynaud’s phenomenon, esophageal symptoms, pulmonary fibrosis, restrictive lung disease, edematous skin, joint contractures, digital ulcers, and muscle weakness. Less frequently occurring, yet life-threatening manifestations include pulmonary artery hypertension (about 1 in 5 patients), cardiac conduction blocks (about 1 in 10 patients), and renal crisis (about 1 in 50 patients). In the US, systemic sclerosis is the most deadly of the systemic autoimmune diseases. A patient with newly diagnosed diffuse cutaneous systemic sclerosis has a 1 in 2 chance of dying by seven years after onset of disease. About 85% of deaths caused by systemic sclerosis are the result of pulmonary fibrosis, pulmonary artery hypertension, or cardiovascular disease, such as sudden death.
In systemic sclerosis the innate immune response fails to transition from the activation phase to the resolution phase. Individuals with systemic sclerosis who have interstitial lung disease have an imbalance of bioactive lipid mediators, causing a predominance of inflammation-activating mediators versus resolving mediators. The preponderance of inflammation-activating mediators correlates positively with the degree of inflammation in the lungs and negatively with forced vital capacity, a measure of lung fibrosis. Excessive activation of the pathways which cause fibrosis including TGFβ, myofibroblast accumulation, and production of collagen and other extracellular matrix proteins are all seen in systemic sclerosis.
Currently, there are no FDA-approved therapies specifically for systemic sclerosis, except secondarily for drugs to treat pulmonary artery hypertension associated with autoimmune diseases. Immunosuppressants with significant toxicities are commonly used to reduce disease activity, generally in the absence of data to support their use. For example, systemic corticosteroids are used frequently in systemic sclerosis patients, despite concerns about toxic side effect and precipitation of renal crisis.
We believe that there is general agreement in the systemic sclerosis community that an effective anti-inflammatory and anti-fibrotic drug would address a significant unmet medical need in systemic sclerosis, especially a drug that is orally administered, can be used chronically with other commonly prescribed medications for systemic sclerosis, and is not immunosuppressive. We believe such a therapy would be positively received by the market.
Dermatomyositis
Dermatomyositis is a serious autoimmune idiopathic inflammatory myopathy with characteristic cutaneous findings. About 30,000 individuals in the U.S. have dermatomyositis. Dermatomyositis usually strikes adults, with most common age of adult onset between 50-60 years.
This systemic disorder most frequently affects the skin and muscles, and dermatomyositis can also include interstitial lung disease/restrictive lung disease, arthritis, gastrointestional and cardiac involvement. Inflammatory muscle disease can cause discomfort and significant weakness of the proximal muscles of the arms and legs and of the trunk. Dermatomyositis can include damaging inflammation elsewhere in the body, for example: lung inflammation that leads to lung fibrosis and restrictive lung disease; heart inflammation that causes arrhythmia, congestive heart failure, and pericarditis, inflammation of muscles in the esophagus that causes swallowing problems or aspiration pneumonia, and arthritis. Dermatomyositis patients may have active skin disease despite successful treatment of their muscle and/or lung disease. The skin findings in dermatomyositis can be disfiguring and are inflammatory rashes characterized by redness and itching in exposed areas of the skin, around the eyes, on the hands, and in a “shawl” distribution on the scalp, hands, upper back, and photo-exposed areas. With this chronic inflammation, patients with dermatomyositis have an increased risk of malignancy, most commonly in older patients By itself, skin involvement in dermatomyositis has a large negative impact on quality of life, comparable to that of cutaneous lupus erythematosus and vulvodynia, and much higher than those of many dermatologic diseases. The pathophysiology of dermatomyositis is also consistent with inability of patients to adequately resolve innate immune responses.
Therapy for dermatomyositis involves both general measures and specific measures to control the muscle disease and the skin disease. In addition, some patients with dermatomyositis need treatment for other systemic manifestations or complications. The muscle component is treated by administering corticosteroids, typically with an immunosuppressive agent. The skin disease is treated by avoiding sun exposure and by using sunscreens and photoprotective clothing, as well as with topical corticosteroids, and antimalarial agents. Antimalarial therapy frequently is ineffective or can cause drug reactions. Antimalarial-refractory disease is then treated with systemic therapies that may additionally cause toxicity, including systemic glucocorticoids, immunosuppressive therapies such as methotrexate, mycophenolate mofetil, or intravenous immunoglobulin.
6
We believe that an effective drug that controls inflammation in the skin, muscles, and other organs will address a significant unmet medical need in dermatomyositis, particularly a drug that is orally administered, can be used chronically with other commonly prescribed medications for the disease, and is not immunosuppressive.
Systemic Lupus Erythematosus
Systemic lupus erythematosus, or SLE or lupus, is a prototypical autoimmune disease with a wide array of clinical manifestations, including arthritis, rash, photosensitivity, oral ulcers, pleuritis, pericarditis, kidney problems, seizures and psychosis and blood cell abnormalities. The musculoskeletal system is the most commonly involved system in SLE. Patients with SLE have an increased frequency of related autoimmune problems, such as Sjogren's syndrome and antiphospholipid syndrome that require additional treatments. Lupus may occur with other autoimmune conditions, such as thyroiditis, hemolytic anemia, and idiopathic thrombocytopenia purpura. Accelerated atherosclerosis among SLE patients is responsible for premature mortality.
The pathology of SLE involves chronic activation of the innate immune system by immune complexes, with activation of complement, increased production of type 1 interferons and other mediators of inflammation and resultant tissue inflammation and damage.
Drugs specifically approved by the FDA for SLE are limited to aspirin, corticosteroids, hydroxychloroquine and belimumab. Physicians commonly treat disease manifestations with immunosuppressive or corticosteroid therapies that have significant toxicities.
We believe that an effective drug that controls inflammation in the joints and skin as well as improves overall disease activity will address a significant unmet medical need in SLE, particularly a drug that is orally administered, can be used chronically with other commonly prescribed medications for the disease, and is not immunosuppressive.
Current Treatment Alternatives for Chronic, Serious Diseases Characterized by Chronic Inflammation and Fibrosis
Overview
Drugs currently used to treat chronic, serious diseases with chronic inflammation and fibrosis are divided broadly into several groups: non-steroidal anti-inflammatory drugs (NSAIDS), anti-malarial agents, systemic corticosteroids, and immunosuppressive agents. The choice of agent or combination generally depends upon the underlying disease and physician and patient preferences.
The potency of NSAIDs in the treatment of chronic, serious diseases with chronic inflammatory and fibrotic diseases is often too limited to control disease activity, and patients may receive additional treatment with anti-malarial drugs, systemic corticosteroids or immunosuppressive agents. Anti-malarial therapy is used as a baseline treatment for chronic inflammation in certain autoimmune diseases, typically SLE and dermatomyositis, especially in patients with milder manifestations of disease. Anti-malarial therapy frequently is ineffective in controlling chronic, serious inflammation, or can cause drug reactions. Antimalarial-refractory disease is then treated with systemic therapies that may additionally cause toxicity, including systemic corticosteroids and immunosuppressive agents.
Systemic corticosteroids are commonly prescribed for treatment of chronic, serious diseases characterized by chronic inflammation and fibrosis, such as cystic fibrosis, systemic sclerosis, and dermatomyositis. Chronic corticosteroid use is limited by toxicities that include growth retardation, iatrogenic Cushings’s Disease, hypertension, high glucose levels/diabetes, obesity, brittle bones/osteoporosis, aseptic necrosis of bone, immunosuppression/increased infection, glaucoma, depression, and psychosis. Thus, safer yet potent alternatives to steroids have long been sought.
Multiple other immunosuppressive drugs are used to treat chronic, serious, inflammatory diseases, to achieve disease control and to reduce or avoid the need for corticosteroids. These include biological agents, such as monoclonal antibodies or fusion proteins, which target a very specific molecule in a key disease pathway. These drugs have a number of disadvantages including that the drugs must be administered by parenterally and they are associated with increased incidence of malignancy and infection. Non-biologic immunosuppressive agents that are used to treat chronic, serious inflammation include methotrexate, mycophenolate, leflunomide, cyclophosphamide, and azathioprine, among others. Intravenous immunoglobulin is used occasionally to treat refractory chronic, serious inflammatory diseases.
Cystic Fibrosis
The importance of treating inflammation in cystic fibrosis is confirmed in the Cystic Fibrosis Foundation’s Strategic Plan, 2014-2018. While treatment with systemic corticosteroids and ibuprofen are effective in improving the symptoms of cystic fibrosis, the side effects associated with chronic treatment using these drugs are significant. Specifically, long term usage of oral corticosteroids in children are associated with glucose intolerance, cataract formation, multiple bone fractures secondary to osteoporosis or osteopenia,
7
Cushing Syndrome effects, and anorexia nervosa as well as growth retardation. The use of high dose ibuprofen is limited by the years of treatment it takes to show benefit, a need to monitor levels closely in the patient, and the increased risk of gastrointestinal bleeding. As a result, these drugs have limited long-term use in cystic fibrosis.
Other therapies routinely used by cystic fibrosis patients include antibiotics, such as Cayston from Gilead and TOBI from Novartis, and mucolytics, such as Pulmozyme from Genentech. In addition, Vertex currently markets the only approved drugs that specifically target the defective CFTR protein, Kalydeco and Orkambi.
Systemic Sclerosis
Cytotoxic medications are used to control overall disease activity in systemic sclerosis. In a study of 2,739 systemic sclerosis patients in the United States, in one year 44.3% received corticosteroids, 4.8% received mycophenolate mofetil, 2.7% received cyclophosphamide, and 0.5% received cyclosporine. In a report of 7,655 patients in the European Scleroderma Trials and Research Group database, immunosuppressant treatments used to treat systemic sclerosis and the percentage of patients receiving them were: prednisone (43.5%) with median dose of 8 mg/day; cyclophosphamide (15.9%); methotrexate (13.7%); azathioprine (6.4%); mycophenolate mofetil (4.2%), d-penicillamine (2.1%), and rituximab (1%).
Dermatomyositis
Current medications for dermatomyositis involve both treatments to reduce overall disease activity and specific treatments to control the muscle disease and the skin disease. The muscle component is treated by administering corticosteroids, typically with an immunosuppressive agent. The skin disease is treated by avoiding sun exposure and by using sunscreens and photoprotective clothing, as well as with topical corticosteroids, antimalarial agents such as hydroxychloroquine and immunosuppressive medications such as methotrexate, azathioprine, mycophenolate mofetil, or intravenous immunoglobulin.
Systemic Lupus Erythematosus
Similar to dermatomyositis, current medications for SLE involve treatments to reduce overall disease activity and specific treatments for a given organ involvement. Commonly used medications include NSAIDs, topical corticosteroids, antimalarial agents, prednisone, belimumab, and other immunosuppressive medications such as mycophenolate, methotrexate, azathioprine, and cyclophosphamide.
Resunab’s Mechanism of Action is Distinct from Anti-Inflammatory Drugs, Steroids and Immunosuppressive Agents
Corticosteroids and NSAIDs exert their effect by inhibiting the activation of inflammation. In simple terms, both classes of drugs inhibit inflammation by “interfering” with the biochemical pathways in the cell that promote and sustain inflammation. For example, NSAIDs directly inhibit the activity of the COX 1 and COX 2 enzymes that are responsible for generating pro-inflammatory eicosanoids. A drawback of this approach is that it only inhibits one arm of the eicosanoid pathway (e.g. COX but not LOX) resulting in an increase in LOX-derived inflammatory mediators which leads to gastrointestinal and cardiovascular side effects (termed “molecular shunting”).
Resunab on the other hand is intended to trigger endogenous pathways that resolve inflammation and halt fibrosis without immunosuppression. Resunab is intended to impact and activate multiple pathways including:
|
|
· |
Increase in production of SPMs and anti-inflammatory eicosanoids, with a concomitant decrease in production of pro-inflammatory eicosanoids. |
|
|
· |
Increase in production of anti-inflammatory cytokines, coupled with a decrease in production of pro-inflammatory cytokines and pro-fibrotic growth factors. |
|
|
· |
Increase in influx of non-inflammatory macrophages with a decrease in influx and accumulation of inflammatory cells and pro-fibrotic myofibroblasts. |
|
|
· |
Increase in bacterial clearance. SPMs stimulate production of bactericidal peptides, enhance phagocytosis and killing of bacteria by neutrophils and macrophages. |
|
|
· |
Increase in apoptosis of inflammatory cells, including neutrophil and pro-fibrotic cells, including fibroblasts. |
|
|
· |
Increase in clearance of apoptotic cells and cellular debris by non-inflammatory macrophages. |
Resunab potentially offers a new and unique mechanism to treat a spectrum of rare, chronic, serious inflammatory and fibrotic diseases.
8
Safety & Toxicology
To date, Resunab has undergone an extensive battery of animal safety and toxicology studies in support of Phase II clinical development. Unlike other CB2 agonists, Resunab exhibits limited blood brain barrier penetration (30%) and negligible CB1 activity in vivo (12.1 binding affinity to CB2 vs. CB1) resulting in limited central nervous system side effects. Results of the safety pharmacology studies in animals assessing the central nervous system, cardiovascular and respiratory systems, renal system and gastrointestinal system were all negative and support that there should be a significant safety margin at anticipated therapeutics doses of Resunab in patients.
Animal toxicological profiles of orally administered Resunab in single and multi-dose studies in mice, rats, and dogs, and a battery of in vitro and in vivo genetic toxicity studies, have been conducted, including a chronic rat toxicology that was requested by the FDA and successfully completed. The results of these studies showed no major toxicological concerns and an excellent safety margin based on drug exposure levels in animals compared to human exposure. In support of Phase II clinical trials, thirteen-week Good Laboratory Practice (“GLP”) toxicology studies have been completed in rats and dogs. Based on the results of these studies and prior clinical trials, the safety margin in humans is estimated to be between 8 and 32 times greater than the doses in our clinical trials.
Pre-clinical Results
In pre-clinical studies, with both prophylactic and therapeutic administration, Resunab has demonstrated clear efficacy at resolving inflammation. Inflammation was induced in animal pharmacology models by a variety of stimuli that trigger acute innate immune responses or mimic established inflammation: arachidonic acid, zymosin, platelet activating factor, IL-1β and TNFα, Freund’s complete adjuvant, and bleomycin. At the histological level, Resunab suppressed inflammation in all of these disease models in which it was studied. At the cellular level, Resunab reduced the numbers of inflammatory cells in the tissue, especially neutrophils. Resunab also induced non-inflammatory death, or apoptosis, of activated T cells. At the molecular level, Resunab stimulated the production of the SPM lipoxin A4 and the anti-inflammatory eicosanoid prostaglandin J2, while reducing production of pro-inflammatory eicosanoids leukotriene B4 and prostaglandin E2. Resunab reduced production of pro-inflammatory cytokines IL-6, TNFα, IL-1β, and type 1 interferons.
Resunab also demonstrated clear efficacy at stopping fibrotic processes in pre-clinical studies, with both prophylactic and therapeutic administration. Fibrosis was induced in joints by Freund’s complete adjuvant, in skin by bleomycin and TGFβ receptor 1 activation, and in lungs by bleomycin. At the histological level, Resunab inhibited the development or progression of fibrosis in each model, with reduced joint ankylisis, skin thickness, and lung fibrosis. For example, at a low dose of 0.1 mg/kg three days a week, Resunab significantly inhibited joint ankylosis (fibrosis) in adjuvant-induced arthritis. At a cellular level, myofibroblast accumulation was reduced (along with inflammatory cell infiltration). At the molecular level, Resunab reduced cellular activation through Erg-1, reduced expression of the pro-fibrotic growth factors TGF-β and connective tissue growth factor, and reduced production of hydroxyproline and collagen.
Collectively, we believe this data supports the development of Resunab as a potent and novel drug to resolve inflammation and halt fibrosis with the potential to treat some of the key manifestations of the indications we are pursuing.
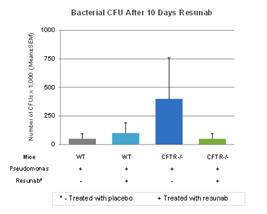
Effect of Resunab in CF Mouse Model
Since patients with CF not only have inflammation, but they are also chronically colonized with bacteria, pre-clinical studies were conducted by Case Western Reserve University to evaluate Resunab’s overall impact on Pseudomonas aeruginosa colonization. Pseudomonas aeruginosa infection was impregnated into wildtype mice (control group) and CFTR-deficient mice in which the CFTR gene was knocked out. Each group then received treatments with Resunab escalating doses 24 hours post infection over a ten day period. At day 10, animals were euthanized and evaluated for bacteria load (colony forming units or “CFUs”), total and differential bronchoalveolar lavage (BAL) white blood cell counts (WBCs). The results of the study demonstrated that in the CFTR-deficient mice group, Resunab improved survival, decreased weight loss, reduced the numbers of neutrophils and white blood cells in the lung and improved the ability of animals to resolve pulmonary infection as assessed by lung bacterial CFUs, compared to control treatment (see Figures 4 & 5 below).
9
Figure 4: Resunab resolves lung inflammation in Pseudomonas Aeruginosa infected mouse model
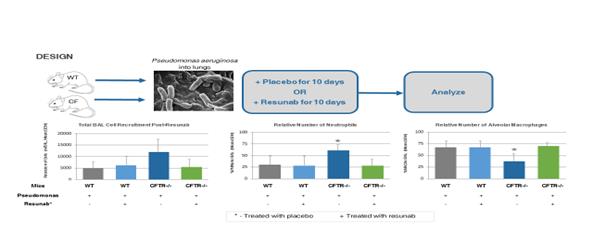
Figure 5: Resunab enhances resolution of lung infection in CF mice infected with Pseudomonas aeruginosa
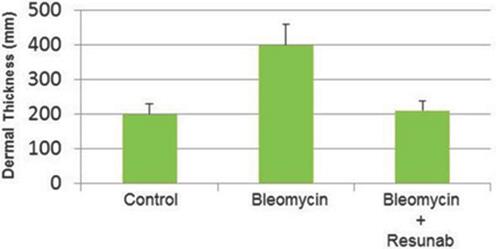
Effect of Resunab in Fibrotic Disease Animal Models
The efficacy of Resunab was investigated in three mouse models of scleroderma. Oral administration of Resunab once-a-day at 1 mg/kg/day inhibited dermal fibrosis in the three models as measured by reductions in dermal thickness, markers of collagen production, and myofibroblast abundance (cells that produce collagen). Histopathologic evaluation revealed that Resunab inhibited the fibrosis of the skin tissue in each of these models. In addition, oral administration of Resunab was effective at inhibiting lung fibrosis in a bleomycin-induced mouse model of systemic sclerosis (Figure 6). Collectively, we believe this data supports the development of Resunab as a potent and novel anti-fibrotic/anti-inflammatory drug with the potential to treat some of the key manifestations of human scleroderma.
10
Figure 6: Resunab inhibits skin thickening in bleomycin-induced model of fibrosis in systemic sclerosis
To translate these in vivo effects of Resunab on fibrosis in animal models to humans, we determined the anti-fibrotic effects of Resunab on dermal fibroblasts isolated from diffuse cutaneous systemic sclerosis patients. Dermal fibroblasts from systemic sclerosis patients overproduce extracellular matrix proteins like collagen and the pro-fibrotic cytokine TGF- β. Further supporting its anti-fibrotic activity, Resunab inhibited new collagen synthesis, reduced TGF-β and increased anti-inflammatory (resolving) eicosanoid levels in this ex-vivo model of human systemic sclerosis. These responses were statistically significant with a p<0.001 as determined using the Student-Newmen Keuls post-hoc test for multiple comparisons. TGF-β has been identified in published scientific literature to be an important cytokine in promoting inflammation and fibrosis in multiple diseases and conditions including systemic sclerosis and cystic fibrosis. While no pre-clinical model is entirely predictive of clinical efficacy, the results from these pre-clinical studies provide a credible rationale for further clinical development.
Human Clinical Results to Date
Two Phase 1 and one Phase 2 clinical trials have been conducted by prior licensees Atlantic Pharmaceuticals and Indevus Pharmaceuticals, Inc. who were developing Resunab as an analgesia therapy (pain relief) rather than an anti-inflammatory therapy. Based on their review of the preclinical and clinical data in pain relief, Indevus Pharmaceuticals elected not to continue with further clinical trials and its license rights were terminated in December 2008. Upon termination, the rights reverted back to Dr. Sumner Burstein who then assigned the rights to us in April 2009.
The first Phase 1 study evaluated the safety, tolerability and pharmacokinetics of a single oral dose of Resunab in healthy adult male subjects over a dose range from 1 mg to 10 mg, in 4 dose groups of 8 subjects each. Resunab had satisfactory oral bioavailability, was well tolerated, and exhibited linear pharmacokinetics over the dose range tested. There were no life-threatening or serious adverse events in this study. Two subjects in the 3 mg group and one subject in the 6 mg group out of a total of twenty-four subjects reported adverse events of mild to moderate intensity. None of the remaining subjects in the 3 mg and 6 mg groups and no subjects in the 1 mg, 10 mg and placebo groups experienced any adverse events. The two subjects in the 3 mg group reported blurred vision, difficulty in remembering, mild euphoria, impression of slower movements, dry mouth and difficulty in concentrating. The one subject in the 6 mg group reported orthostatic vagal fainting, feeling dizzy, and nausea immediately after the first blood draw, which occurred prior to the administration of the drug.
The second Phase 1 study evaluated the safety, tolerability and pharmacokinetics of single and multiple ascending doses of Resunab at higher doses. Each dose level had 8 subjects randomized at a ratio of 6 receiving Resunab and 2 receiving placebo. Doses ranged from 10 to 240 mg single dose, and 20, 40 and 80 mg three times a day, or tid, for 7 days multi-dose. Resunab showed good tolerability at single doses up to 120 mg and multiple doses up to 40 mg three times a day for 7 days. For the single dose treatment stage, the most frequently occurring treatment–emergent adverse events, or TEAEs, occurring in > 10% of all subjects treated with Resunab, in decreasing order of frequency, were: dizziness (15 out of 48), nausea (10 out of 48), vomiting (7 out of 48), pallor (6 out of 48), dry mouth (5 out of 48), headache (5 out of 48), somnolence (5 out of 48), tremor (5 out of 48), and disorientation (5 out of 48). All TEAEs were mild to moderate and the majority of these events occurred in subjects treated at dose levels of 120 mg and above. For the multiple dose stage, there were no TEAEs at 20 mg tid while in the 40 and 80 mg tid dose groups the most frequently occurring TEAEs, occurring in > 10% of all subjects treated with Resunab, were at the 40 and 80 mg tid dose respectively: dizziness (4 out of 6 and 3 out of 6), nausea (2 out of 6 and 1 out of 6), somnolence (2 out of 6 and 1 out of 6), dry mouth (1 out of 6 and 1 out of 6), fatigue (0 out of 6 and 2 out of 6), feeling abnormal (0 out of 6 and 2 out of 6), anorexia (0 out of 6 and 2 out of 6),
11
inappropriate affect (0 out of 6 and 2 out of 6), and orthostatic hypotension (0 out of 6 and 2 out of 6). The majority of these events were mild and occurred in subjects treated at the highest dose level of approximately 80 mg three times a day, for 7 days (i.e., subjects treated with 80 mg tid were reduced to 60 mg tid starting with the second dose on Day 2 continuing through Day 7). In some elderly patients over 65 years of age, changes in electrocardiogram readings were noted; however no differences in readings were observed between subjects treated with Resunab versus and placebo thus this was not deemed to be of clinical relevance. Resunab exhibited linear pharmacokinetics over the dose range tested.
A Phase 2 crossover design study was conducted for Resunab in refractory neuropathic pain patients, or NP. In this study, 21 NP patients were treated with either Resunab or placebo at a dose of 20 and 40 mg twice a day (8 hours apart) for 1 week followed by a 1 week washout and a cross over. Resunab reduced the 3 hour pain index by up to 28% (p<0.03) in one cohort, and was effective at reducing pain by >30% in 50% of this same cohort (P<0.03) of refractory pain patients although its analgesic effect wore off between 3-8 hours post-dose. Resunab was well tolerated and there were no safety issues of note in this study.
While Resunab showed a clear analgesic signal in this phase 2 study in refractory neuropathic pain patients, we are not relying on these efficacy results to support our clinical program as this study was designed to measure pain while our future studies will have a completely different efficacy endpoint.
Clinical Development Plan
Overview
We are currently conducting Phase 2 randomized, double-blind, placebo-controlled clinical trials in cystic fibrosis, systemic sclerosis, and dermatomyositis. These studies are evaluating the safety, efficacy, and pharmacokinetics of Resunab in patients, during 84 days of active treatment. Proof of CB2-mediated biologic activity is being tested with metabolipidomic profiles, testing for shift in production of bioactive lipid mediators to SPMs and away from inflammation-activating lipid mediators. Evidence of downstream effects of Resunab on relevant pathologic processes is being evaluated using biomarkers of inflammation and disease activity in blood and involved tissues. In addition we plan to commence a clinical study during the first quarter of 2017 in Systemic Lupus Erythematosus, or SLE.
Upon obtaining adequate safety, efficacy, and mechanistic data to support selection of outcome, dose, and sample size in pivotal clinical studies in a given indication, we plan to conduct pivotal, global clinical trial(s) in cystic fibrosis, systemic sclerosis, and/or dermatomyositis. We will seek input and concordance from regulatory authorities on the design of these pivotal studies.
We have obtained Orphan Drug Designations and Fast Track status for both CF and systemic sclerosis. Fast Track designation is eligible for some or all of the following: (i) more frequent meetings with FDA to discuss the drug’s development plan and ensure collection of appropriate data needed to support drug approval; (ii) more frequent written correspondence from FDA about things such as the design of the proposed clinical trial and the use of biomarkers; and (iii) eligibility for Accelerated Approval and Priority Review, if relevant criteria are met.
Cystic Fibrosis
In September 2015, we initiated a Phase 2 study in Resunab in patients suffering from CF. The study is a multi-center U.S. and European trial. The lead Principal Investigator in the U.S. is Dr. James Chmiel of the Rainbow Babies & Children’s Hospital, Cleveland, Ohio and the Co-Investigator in Europe is Dr. Stuart Elborn of Queens University Hospital, Belfast, Ireland.
The primary objective of the CF study is to evaluate safety and tolerability of Resunab in seventy adults with cystic fibrosis. Secondary objectives are to assess Resunab concentrations and to evaluate efficacy of Resunab in cystic fibrosis. Cystic fibrosis patients in the study are between 18-70 years of age with forced expiratory volume in one second ≥ 40% predicted. Subjects are permitted to remain on their baseline medications for cystic fibrosis and do not need to have a specific mutation of the CFTR. The trial has screening period of 28 days, followed by 112 days of active treatment, and then 28 days safety follow-up. Resunab is being evaluated at initial dosing levels of 1 mg and 5 mg per day for the first 28 days, and then 20 mg once a day and 20 mg twice a day for the next 56 days, versus placebo. Patients are being monitored for safety and tolerability throughout the study. Safety will be assessed with physical examinations, adverse effects, laboratory safety testing, electrocardiograms and testing of psychoactivity. Other assessments will include Resunab concentrations and metabolites measurements of lung function, patient-reported outcomes, and biomarkers of inflammation in the blood and sputum, and metabolipidomic profiles. The Company expects to complete the study and report data by the end of 2016.
12
Systemic Sclerosis (Scleroderma)
In August 2015, we initiated a Phase 2 double-blind placebo-controlled clinical study in patients suffering from diffuse cutaneous systemic sclerosis. The study is a U.S. multi-center trial. The Principal Investigator is Dr. Robert Spiera of the Hospital of Special Surgery, New York City, New York.
Patients in the systemic sclerosis trial have disease duration less than six years and are permitted to remain on their baseline mediations. Resunab is being evaluated at three dosing levels - 5 mg, 20 mg once a day, and 20 mg twice a day - versus placebo. The trial has a 28 day screening period, followed by 112 days of active treatment period and then a 28 day safety follow-up. The primary goals of this trial are to evaluate safety and explore efficacy of Resunab in thirty six patients with diffuse systemic sclerosis. Patients are being monitored for safety and tolerability throughout the study. Safety is being assessed with physical examinations, adverse effects, laboratory safety testing, electrocardiograms and testing of psychoactivity. Efficacy is being assessed using the Combined Response Index in diffuse cutaneous Systemic Sclerosis and its domains that measure skin thickening, lung function, physician and patient global assessments of general health, and disability. Secondary endpoint measurements include Resunab concentrations and metabolites, other patient-reported outcomes, biomarkers of inflammation and fibrosis in the blood and skin, and metabolipidomic profiles. We expect to complete the study and report data by the end of 2016.
Dermatomyositis
In June 2015, we initiated a Phase 2 double-blind placebo-controlled clinical study evaluating Resunab in patients with skin-predominant dermatomyositis. The study is a U.S. single center trial and is being funded by a N.IH grant. The Principal Investigator is Dr. Victoria Werth of the University of Pennsylvania, Philadelphia, PA.
Patients in the dermatomyositis study have active skin involvement, relatively inactive muscle disease, and are permitted to remain on their baseline mediations. Resunab is being evaluated at two dosing levels - 20 mg once a day and 20 mg twice a day - versus placebo. The trial design has a 28 day screening period, followed by 112 days treatment period and a 28 day safety follow-up. The primary goals of this trial are to evaluate safety and efficacy of Resunab in 22 patients with skin-predominant dermatomyositis. Patients are being monitored for safety and tolerability throughout the study. Safety is being assessed with physical examinations, adverse effects, laboratory safety testing, electrocardiograms and testing of psychoactivity. Efficacy is being assessed using the Cutaneous Dermatomyositis Disease Area and Severity Index. Secondary endpoint measurements include Resunab concentrations, other patient-reported outcomes, biomarkers of inflammation and disease activity in the blood and skin, and metabolipidomic profiles. Data from this single center study is expected Q1 2017.
Systemic Lupus Erythematosus
We plan to commence a Phase 2 double blind placebo-controlled study evaluating Resunab in the treatment of SLE in the first quarter of 2017. The study is being funded by an NIH Grant to the Feinstein Institute for Medical Research and will test the efficacy, safety, tolerability and biologic effects of Resunab as a novel, non-immunosuppressive oral treatment to improve signs and symptoms of SLE. The study plans to enroll about 100 adult SLE patients with active musculoskeletal disease and will be carried out at approximately 10 sites in the United States. These patients will receive either placebo or three different doses of Resunab daily for 84 days with 28 days follow-up.
Competition
There are numerous drug therapies currently used to treat CF patients, targeting different aspects of this complex disease. Inhaled and oral antibiotics address the pulmonary microbial infection. Mucolytics address the accumulation of mucus in the lungs. Bronchodilators and hydration agents are also used to help improve pulmonary function. Targeting of the inflammatory component of the disease is currently done by high dose Ibuprofen and oral corticosteroids. While these offer some clinical benefit, they are not used chronically due to their adverse side effects which include immunosuppression and metabolic changes (steroids) as well as the risk of gastrointestinal bleeding (ibuprofen). Thus, there is a clear and urgent unmet medical need for safe and effective inflammation-targeting drugs for the chronic treatment of CF that could potentially have a beneficial impact on morbidity and mortality.
An emerging area of CF therapy is the development of correctors and potentiators of CFTR. In January 2012, Vertex launched Kalydeco™, or ivacaftor, the first ever cystic fibrosis drug specifically targeting the underlying genetic defect in the CFTR ion channel. Kalydeco is a small synthetic oral molecule that helps potentiate the function of the G551D mutant CFTR protein, resulting in improved forced expiratory volume in one second (a measure of obstruction of airflow in the lungs) by approximately 10% in cystic fibrosis patients. With at least 1,800 different known mutations in the CFTR gene, Kalydeco can only be used by a specific sub-section of the cystic fibrosis population (ages 2 and above) who suffer from a limited number of “gating mutations,” including the G551D mutation. Combined, these currently account for about 5.7% of the total cystic fibrosis worldwide patient population or about 4,000 individuals.
13
A new combination drug from Vertex, Orkambi™ (lumacaftor/ivacaftor) combination treatment targets a larger population of homozygote ΔF508 CFTR mutation patients. Orkambi was approved by the FDA on July 2, 2015. In clinical studies, the lung function of patients taking Orkambi improved by a range of 2.6 percentage points to 3 percentage points, compared with that of patients receiving placebo. Initially, Orkambi will target the approximately 8,500 homozygotic patients carrying two copies of the ΔF508 mutation, but further approval may be sought for heterozygotic patients (one copy of the ΔF508 and another copy of a different CFTR mutation). Together these two patient populations are estimated to be about 70% of cystic fibrosis patients.
Several other companies are developing drugs for CF targeting CFTR either as a protein or mRNA transcript. These are highlighted in the table below:
|
Selected CF Products in Development |
|||||||
|
Company |
Drug |
Mechanism |
Delivery |
Mutation |
Stage |
||
|
PTC Therapeutics |
Ataluren |
Ribosome read thru (nonsense mutations) |
Oral |
Class 1, nonsense |
Phase 3 |
||
|
UK CF Gene Therapy Consortium |
pGM169/GL67A |
Gene therapy |
Inhaled |
All |
Phase 2b |
||
|
Novartis |
QBW251 |
Potentiator |
Oral |
Gating |
Phase 2 |
||
|
Bayer |
Riociguat |
stimulates sGC enzyme |
Oral |
F508del homozygous |
Phase 2 |
||
|
Nivalis Therapeutics (formerly N30) |
N91115 |
GSNOR inhibitor |
IV and Oral |
F508del homozygous |
Phase 1b |
||
|
Flatley Discovery Lab |
FDL169 |
Corrector |
Oral |
F508del |
Phase 1 |
||
|
Galapagos / AbbVie |
GLPG1837 / ABBV-974 |
Potentiator |
Oral |
Gating |
Phase 1 |
||
|
ProQR |
QR-010 |
RNA oligonucleotide |
Inhaled |
F508del homozygous |
Phase 1 |
||
|
Celtaxsys |
Acebilustat |
Anti-inflammatory-inhibits production of LTB4 |
Oral |
N/A |
Phase 2 |
||
|
Proteostasis |
PTI130 |
CFTR amplifier |
Oral |
All |
Preclinical |
||
For autoimmune disorders such as systemic sclerosis, dermatomyositis and SLE, physicians treat patients with a number of drugs including potent immunosuppressants and cytotoxics to try to reduce the autoimmune response characteristic of the disease. These drugs have not proven to be very effective thus there remains a high unmet need for safe and effective drugs to treat these autoimmune disorders. Several companies, including Roche, Boehringer Ingelheim, Bristol Myers, Sanofi, Promedior and Digna Biotech, are actively working to develop new drugs for treating the inflammation and/or fibrosis in systemic sclerosis. To the best of our knowledge, Resunab offers a unique mode of action to treat systemic sclerosis being one of the few oral drugs with the potential to resolve inflammation and halt fibrosis without immunosuppression.
Research and Development
We incurred expenses of approximately $5,889,000 and $1,256,000 for research and development activities for the years ended December 31, 2015 and 2014, respectively. These expenses include cash and non-cash expenses relating to the development of our clinical and pre-clinical programs for Resunab.
14
Intellectual Property
Our intellectual property and product pipeline is based on seminal research done by Professor Sumner Burstein, Ph.D. at the University of Massachusetts Medical Center and patents filed by Corbus directly. Upon Corbus’ formation, Dr. Burstein assigned to Corbus his rights and intellectual property ownership to Resunab on a royalty-free basis in consideration for 200,000 shares of Series A Non-Convertible preferred stock. The intellectual property includes scientific data and regulatory data, product know-how and one patent issued in January 2014 and expiring in November 2029 for the use of Resunab to treat interstitial cystitis, an indication for which we are currently pursuing partnership opportunities.
Since acquiring the asset, we have filed three new patent applications for Resunab, which if granted, would extend intellectual property protection through at least 2033. The three new patent filings cover:
|
|
· |
A composition of matter claim based on an improved ultrapure version of Resunab; |
|
|
· |
Resunab’s use in treating a number of pulmonary fibrotic diseases including cystic fibrosis, systemic sclerosis and idiopathic pulmonary fibrosis; and others |
|
|
· |
Specific Resunab dosing regimens and formulations. |
The FDA has granted Resunab an Orphan Drug Designation for both cystic fibrosis and systemic sclerosis and we will be seeking orphan drug status for these indications in Europe. For dermatomyositis and possibly other orphan inflammatory diseases we will be seeking orphan drug status from the FDA and in Europe. Orphan drug status provides seven years of market exclusivity in the U.S. and ten years in Europe beginning on the date of drug approval.
Our commercial success depends in part on our ability to obtain and maintain patent and other proprietary protection for Resunab and to operate without infringing the proprietary right of others and to prevent others from infringing our proprietary rights. We strive to protect our intellectual property through a combination of patents, and trademarks as well as through the confidentiality provisions in our contracts. With respect to Resunab, we endeavor to obtain and maintain patent protection in the United States and internationally on all patentable aspects of the drug. We cannot be sure that the patents will be granted with respect to any patent applications we may own or license in the future, nor can we be sure that our existing patents or any patents we may own or license in the future will be useful in protecting our technology. For this and more comprehensive risks related to our intellectual property, please see “Risk Factors—Risks Relating to Our Intellectual Property Rights.”
In addition to patents, we rely on trade secrets and know-how to develop and maintain our competitive position. For example, significant aspects of our proprietary technology platform are based on unpatented trade secrets and know-how. Trade secrets and know-how can be difficult to protect. We seek to protect our proprietary technology and processes, in part, by confidentiality agreements and invention assignment agreements with our employees, consultants, scientific advisors, contractors and commercial partners. These agreements are designed to protect our proprietary information and, in the case of the invention assignment agreements, to grant us ownership of technologies that are developed through a relationship with a third party. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, agreements or security measures may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our contractors use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
We also plan to seek trademark protection in the United States and outside of the United States where available and when appropriate. We intend to use these registered marks in connection with our pharmaceutical research and development as well as our product candidates.
Manufacturing and Supply for Resunab
We have developed and validated a good manufacturing practice, or GMP, process to manufacture Resunab API and drug product through our contract manufacturers. The API contract manufacturer has already produced multi-Kg scale bulk batches of the active pharmaceutical ingredient under GMP for our Phase 2 clinical studies and is capable of producing sufficient active ingredient for all the clinical studies required prior to submitting an NDA filing with the FDA. We do not own or operate manufacturing facilities for the production of Resunab. We expect to depend on third-party suppliers and manufacturing organizations for all of our clinical trial quantities of raw materials and drug substance. Resunab is a synthetic molecule and there are readily available supplies of all raw materials needed for the manufacture of Resunab.
15
Regulatory Matters
Government Regulation
Any product development activities related to Resunab or products that we may develop or acquire in the future will be subject to extensive regulation by various government authorities, including the FDA and other federal, state and local statutes and regulations and comparable regulatory authorities in other countries, which regulate the design, research, clinical and non-clinical development, testing, manufacturing, storage, distribution, import, export, labeling, advertising and marketing of pharmaceutical products and devices. Generally, before a new drug can be sold, considerable data demonstrating its quality, safety and efficacy must be obtained, organized into a format specific to each regulatory authority, submitted for review and approved by the regulatory authority. The data are often generated in two distinct development states: pre-clinical and clinical. Resunab or other products that we may develop or acquire in the future must be approved by the FDA through the IND process before they may be legally marketed in the United States. For new chemical entities, the pre-clinical development stage generally involves synthesizing the active component, developing the formulation and determining the manufacturing process, as well as carrying out non-human toxicology, pharmacology and drug metabolism studies which support subsequent clinical testing.
The clinical stage of development can generally be divided into three sequential phases that may overlap, Phase 1, Phase 2 and Phase 3 clinical trials. In Phase 1, generally, small numbers of healthy volunteers are initially exposed to single escalating doses and then multiple escalating doses of the product candidate. The primary purpose of these studies is to assess the metabolism, pharmacologic action, side effect tolerability and safety of the drug. Phase 2 trials typically involve studies in disease-affected patients to determine the dose required to produce the desired benefits, while Phase 2b trials are designed to determine efficacy. At the same time, safety and further pharmacokinetic and pharmacodynamic information is collected. In some instances, formal Phase 1 and Phase 2 trials may not be deemed necessary or required by the FDA. Such is often the case when the safety and efficacy of an active ingredient is considered to be well understood by the FDA. Under established regulatory frameworks, pharmaceutical products with active ingredients equal or similar to those known by the FDA often enter more streamlined development programs than compounds entirely new to the agency.
Post-approval studies, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. Sometimes, these studies are used to gain additional experience from the treatment of patients in the intended therapeutic condition, then often referred to as Phase 4 clinical trials. In certain instances, the FDA may mandate the performance of Phase 4 studies. In other situations, post-approval studies aim to gain additional indications for a medication..
Development of Drugs in the United States
In the United States, the process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable United States requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning letters, product recalls or withdrawals from the market, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement, or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
Prior to the start of human clinical studies for a new drug in the United States, pre-clinical laboratory and animal tests are often performed under the FDA’s Good Laboratory Practices regulations. The sponsor must submit the result of the pre-clinical tests, together with manufacturing information, analytical data and any available clinical data or literature and a proposed clinical protocol to the FDA as part of an IND application, which is a request for authorization from the FDA to administer an investigational drug or biological product to humans. Similar filings are required in other countries. The amount of data that must be supplied in the IND depends on the phase of the study. Phase 1 studies typically require less data than larger Phase 2 and 3 studies. A clinical plan must be submitted to the FDA prior to commencement of a clinical trial. If the FDA has concerns about the clinical plan or the safety of the proposed studies, they may suspend or terminate the study at any time. Studies must be conducted in accordance with good clinical practice and regulator reporting of study progress and any adverse experiences is required. Studies are also subject to review by independent institutional review boards responsible for overseeing studies at particular sites and protecting human research study subjects. An independent institutional review board may also suspend or terminate a study once initiated. Accordingly, we cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin, or that once begun, issues will not arise that could cause the trial to be suspended or terminated.
Review and Approval in the United States
Following pivotal or Phase 3 trial completion, data are analyzed to determine safety and efficacy. Data are then filed with the FDA in a New Drug Application, or an NDA, along with proposed labeling for the product and information about the manufacturing
16
and testing processes and facilities that will be used to ensure product quality. In the United States, FDA approval of an NDA must be obtained before marketing a pharmaceutical product. The NDA must contain proof of safety, purity, potency and efficacy, which entails extensive pre-clinical and clinical testing.
The FDA will likely re-analyze the clinical trial data, which could result in extensive discussions between the FDA and us during the review process. The review and evaluation of applications by the FDA is extensive and time consuming and may take several years to complete. The FDA may conduct a pre-approval inspection of the manufacturing facilities for the new product to determine whether they comply with current good manufacturing practice requirements and may also audit data from clinical and pre-clinical trials.
There is no assurance that the FDA will act favorably or quickly in making such reviews and significant difficulties or costs may be encountered in our efforts to obtain FDA approvals. The FDA may require that certain contraindications, warnings or precautions be including in the product labeling, or may condition the approval of the NDA on other changes to the proposed labeling, development of adequate controls and specifications, or a commitment to conduct post-marketing testing or clinical trials and surveillance programs to monitor the safety of approved products that have been commercialized. Further, the FDA may place conditions on approvals including the requirement for a risk evaluation and mitigation strategy, or REMS, to assure the safe use of the drug. If the FDA concludes a REMS is needed, the sponsor of the NDA must submit a proposed REMS; the FDA will not approve the NDA without an approved REMS, if required. A REMS could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of products. Product approvals maybe withdrawn for non-compliance with regulatory standards or if problems occur, following the initial marketing of the product.
Orphan Drug Designation
Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making a drug available in the Unites States for this type of disease or condition will be recovered from sales of the product. Orphan product designation must be requested before submitting an NDA. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan product designation does not convey any advantage in or shorten the duration of regulatory review and approval process. In addition to the potential period of exclusivity, orphan designation makes a company eligible for grant funding of up to $400,000 per year for four years to defray costs of clinical trial expenses, tax credits for clinical research expenses and potential exemption from the FDA application user fee.
If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan drug exclusivity, which means the FDA may not approve any other applications to market the same drug for the same indication for seven years, except in limited circumstances, such as (i) the drug’s orphan designation is revoked; (ii) its marketing approval is withdrawn; (iii) the orphan exclusivity holder consents to the approval of another applicant’s product; (iv) the orphan exclusivity holder is unable to assure the availability of a sufficient quantity of drug; or (v) a showing of clinical superiority to the product with orphan exclusivity by a competitor product. If a drug designated as an orphan product receives marketing approval for an indication broader than what is designated, it may not be entitled to orphan drug exclusivity. There can be no assurance that we will receive orphan drug designation for Resunab in the indications of cystic fibrosis, systemic sclerosis, or other orphan inflammatory diseases.
Drug Development in Europe
In the European Union, our future products may also be subject to extensive regulatory requirements. Similar to the United States, the marketing of medicinal products has been subject to the granting of marketing authorizations by regulatory agencies. Particular emphasis is also being placed on more sophisticated and faster procedures for reporting of adverse events to the competent authorities.
As in the United States, the various phases of pre-clinical and clinical research in the European Union are subject to significant regulatory controls. Although the regulatory controls on clinical research are currently undergoing a harmonization process following the adoption of the Clinical Trials Directive 2001/20/EC, there are currently significant variations in the member state regimes. All member states, however, currently require independent institutional review board approval of interventional clinical trials. Except for the United Kingdom Phase 1 studies in health volunteers, all clinical trials require either prior governmental notification or approval. Most regulators also require the submission of adverse event reports during a study and a copy of the final study report.
17
Review and Approval in the European Union
In the European Union, approval of new medicinal products can be obtained through one of three processes: the mutual recognition procedure, the centralized procedure and the decentralized procedure. We intend to determine which process we will follow, if any, in the future.
Mutual Recognition Procedure: An applicant submits an application in one European Union member state, known as the reference member state. Once the reference member state has granted the marketing authorization, the applicant may choose to submit applications in other concerned member states, requesting them to mutually recognize the marketing authorizations already granted. Under this mutual recognition process, authorities in other concerned member states have 55 days to raise objections, which must then be resolved by discussion among the concerned member states, the reference member state and the applicant within 90 days of the commencement of the mutual recognition procedure. If any disagreement remains, all considerations by authorities in the concerned member states are suspended and the disagreement is resolved through an arbitration process. The mutual recognition procedure results in separate national marketing authorizations in the reference member state.
Centralized Procedure: This procedure is currently mandatory for products developed by means of a biotechnological process and optional for new active substances and other “innovative medicinal products with novel characteristics.” Under this procedure, an application is submitted to the European Agency for the Evaluation of Medical Products. Two European Union member states are appointed to conduct an initial evaluation of each application. These countries each prepare an assessment report that is then used as the basis of a scientific opinion of the Committee on Proprietary Medical Products. If this opinion is favorable, it is sent to the European Commission, which drafts a decision. After consulting with the member states, the European Commission adopts a decision and grants a marketing authorization, which is valid throughout the European Union and confers the same rights and obligations in each of the member states as a marketing authorization granted by that member state.
Decentralized Procedure: The most recently introduced of the three processes for obtaining approval of new medicinal processes in the European Union, the decentralized procedure is similar to the mutual recognition procedure described above, but with differences in the timing that key documents are provided to concerned member states by the reference member state, the overall timing of the procedure and the possibility of, among other things, “clock stops” during the procedure.
Post-Marketing Requirements
Following approval of a new product, a pharmaceutical company and the approved product are subject to continuing regulation by the FDA and other federal and state regulatory authorities, including, among other things, monitoring and recordkeeping activities, reporting to applicable regulatory authorities of adverse experiences with the product, providing the regulatory authorities with updated safety and efficacy information, product sampling and distribution requirements, and complying with promotion and advertising requirements, which include, among others, standards for direct-to-consumer advertising, restrictions on promoting drugs for uses or in patient populations not described in the drug’s approved labeling (known as “off-label use”), limitations on industry-sponsored scientific and educational activities, and requirements for promotional activities involving the internet. Although physicians may prescribe legally available drugs for off-label uses, manufacturers may not market or promote such off-label uses. Modifications or enhancements to the products or labeling or changes of site of manufacture are often subject to the approval of the FDA and other regulators, which may or may not be received or may result in a lengthy review process.
Prescription drug advertising is subject to federal, state and foreign regulations. In the United States, the FDA regulates prescription drug promotion, including direct-to-consumer advertising. Prescription drug promotion materials must be submitted to the FDA in conjunction with their first use. Any distribution of prescription drug products and pharmaceutical samples must comply with the U.S. Prescription Drug Marketing Act, a part of the U.S. Federal Food, Drug and Cosmetic Act. Once a product is approved, its manufacture is subject to comprehensive and continuing regulations by the FDA. The FDA regulations require the products be manufactured in specific approved facilities and in accordance with current good manufacturing practices, and NDA holders must list their products and register their manufacturing establishments with the FDA. These regulations also impose certain organizational, procedural and documentation requirements with respect to manufacturing and quality assurance activities. Drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with current good manufacturing practice and other laws. NDA holders using contract manufacturers, laboratories or packagers are responsible for the selection and monitoring of qualified firms. These firms are subject to inspections by the FDA at any time, and the discovery of violative conditions could result in enforcement actions that interrupt the operation of any such facilities or the ability to distribute products manufactured, processed or tested by them.
Special Protocol Assessment
The Federal Food, Drug and Cosmetic Act directs the FDA to meet with sponsors, pursuant to a sponsor’s written request, for the purpose of reaching agreement on the design and size of clinical trials intended to form the primary basis of an efficacy claim in an
18
NDA. If an agreement is reached, the FDA will reduce the agreement to writing and make it part of the administrative record. This agreement is called a special protocol assessment, or SPA. While the FDA’s guidance on SPAs states that documented SPAs should be considered binding on the review division, the FDA has latitude to change its assessment if certain exceptions apply. Exceptions include public health concerns emerging that were unrecognized at the time of the protocol assessment, identification of a substantial scientific issue essential to the safety or efficacy testing that later comes to light, a sponsor’s failure to follow the protocol agreed upon, or the FDA’s reliance on data, assumptions or information that are determined to be wrong.
Other Regulatory Matters
Manufacturing, sales, promotion and other activities following product approval are also subject to regulation by numerous regulatory authorities in addition to the FDA, including, in the United States, the Centers for Medicare & Medicaid Services, or CMS, other divisions of the Department of Health and Human Services, the Drug Enforcement Administration, the Consumer Product Safety Commission, the Federal Trade Commission, the Occupational Safety & Health Administration, the Environmental Protection Agency, and state and local governments. Sales, marketing and scientific/educational programs must also comply with federal and state fraud and abuse laws. Pricing and rebate programs must comply with the Medicaid rebate requirements of the U.S. Omnibus Budget Reconciliation Act of 1990. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. The handling of any controlled substances must comply with the U.S. Controlled Substances Act and Controlled Substances Import and Export Act. Products must meet applicable child-resistant packaging requirements under the U.S. Poison Prevention Packaging Act. Manufacturing, sales, promotion and other activities are also potentially subject to federal and state consumer protection and unfair completion laws.
In the United States, our product candidate, Resunab, is currently classified as Schedule I controlled substance as defined in the Controlled Substance Act (“CSA”). This designation is based on Resunab’s chemical structure and pharmacology (namely, it being a synthetic endocannabinoid mimetic that binds to the CB2 receptor). Even though Resunab’ s mechanism of action is to modulate the immune system and results to date from clinical studies have demonstrated the drug has no psychotropic effects (which we believe is unlike other members of its chemical class), the DEA classifies Resunab as a Schedule I substance.
Schedule I controlled substances are pharmaceutical products subject to specific regulations under the CSA, that establishes, among other things, certain registration, manufacturing quotas, security, recordkeeping, reporting, import, export and other requirements administered by the DEA. All parties responsible for the manufacturing, distribution and testing the drug in clinical studies must apply for and obtain a license from the DEA before they are permitted to perform these activities with Resunab. Furthermore, these parties must have the security, control, recordkeeping, reporting and inventory mechanisms required by the DEA to prevent drug loss and diversion. All licensed facilities are required to renew their registrations annually if they intend to continue to work with our drug. The DEA conducts periodic inspections of certain registered establishments that handle controlled substances. We have been working with our manufacturers, distributors, exporters and clinical sites to obtain the necessary licenses to work with Resunab. The parties responsible for the manufacturing, distribution and export of Resunab have already applied for and have been granted DEA licenses and a number of institutions responsible for conducting our Phase 2 clinical studies have also been granted DEA licenses
Individual states have also established controlled substance laws and regulations. Though state-controlled substances laws often mirror federal law, because the states are separate jurisdictions, they may separately schedule drugs, as well. While some states automatically schedule a drug based on federal action, other states schedule drugs through rulemaking or a legislative action. The requirement for state registrations could also result in delay of the manufacturing, distribution of Resunab or in the completion of the Phase 2 clinical studies. We and our manufacturing vendors and clinical sites must also obtain separate state registrations, permits or licenses in order to be able to obtain, handle, and distribute controlled substances for clinical trials or commercial sale, and failure to meet applicable regulatory requirements could lead to enforcement and sanctions by the states in addition to those from the DEA or otherwise arising under federal law.
The distribution of pharmaceutical products is subject to additional requirements and regulations, including extensive record-keeping, licensing, storage and security requirements intended to prevent the unauthorized sale of pharmaceutical products.
Third-Party Payer Coverage and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any of our drug candidates that ultimately may obtain regulatory approval. In both the United States and foreign markets, our ability to commercialize our product candidates successfully, and to attract commercialization partners for our product candidates, depends in significant part on the availability of adequate financial coverage and reimbursement from third-party payers, including, in the United States, governmental payers such as the Medicare and Medicaid programs, managed care organizations, and private health insurers. Medicare is a federally funded program managed by the CMS, through local fiscal intermediaries and carriers that administer coverage and reimbursement for certain healthcare items and services furnished to the elderly and disabled. Medicaid is an insurance program for certain categories of patients
19
whose income and assets fall below state defined levels and who are otherwise uninsured that is both federally and state funded and managed by each state. The federal government sets general guidelines for Medicaid and each state creates specific regulations that govern its individual program. Each payer has its own process and standards for determining whether it will cover and reimburse a procedure or particular product. Private payers often rely on the lead of the governmental payers in rendering coverage and reimbursement determinations. Therefore, achieving favorable CMS coverage and reimbursement is usually a significant gating issue for successful introduction of a new product. The competitive position of some of our products will depend, in part, upon the extent of coverage and adequate reimbursement for such products and for the procedures in which such products are used. Prices at which we or our customers seek reimbursement for our product candidates can be subject to challenge, reduction or denial by the government and other payers.
The United States Congress and state legislatures may, from time to time, propose and adopt initiatives aimed at cost containment, which could impact our ability to sell our product candidates profitably. For example, in March 2010, President Obama signed into law the Patient Protection and Affordable Care Act and the associated reconciliation bill, which we refer to collectively as the Health Care Reform Law, a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. Effective October 1, 2010, the Health Care Reform Law revised the definition of “average manufacturer price” for reporting purposes, which could increase the amount of Medicaid drug rebates to states once the provision is effective. Further, the law imposes a significant annual fee on companies that manufacture or import branded prescription drug products. Substantial new provisions affecting compliance have also been enacted, which may require us to modify our business practices with healthcare practitioners. We will not know the full effects of the Health Care Reform Law until applicable federal and state agencies issue regulations or guidance under the new law. Although it is too early to determine the effect of the Health Care Reform Law, the new law appears likely to continue the pressure on pharmaceutical pricing, especially under the Medicare program, and may also increase our regulatory burdens and operating costs. Moreover, in the coming years, additional changes could be made to governmental healthcare programs that could significantly impact the success of our product candidates.
The cost of pharmaceuticals continues to generate substantial governmental and third-party payer interest. We expect that the pharmaceutical industry will experience pricing pressures due to the trend toward managed healthcare, the increasing influence of managed care organizations and additional legislative proposals. Our results of operations could be adversely affected by current and future healthcare reforms.
Some third-party payers also require pre-approval of coverage for new or innovative devices or drug therapies before they will reimburse healthcare providers that use such therapies. While we cannot predict whether any proposed cost-containment measures will be adopted or otherwise implemented in the future, the announcement or adoption of these proposals could have a material adverse effect on our ability to obtain adequate prices for our product candidates and operate profitably.
In addition, in some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products. Historically, products launched in the European Union do not follow price structures of the United States and generally tend to be significantly lower.
20
Other Healthcare Laws and Compliance Requirements
In the United States, our activities are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including the CMS, other divisions of the United States Department of Health and Human Services (e.g., the Office of Inspector General), the United States Department of Justice and individual United States Attorney offices within the Department of Justice, and state and local governments. These regulations include:
|
|
· |
the federal healthcare program anti-kickback law which prohibits, among other things, persons from soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual, for an item or service or the purchasing or ordering of a good or service, for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs; |
|
|
· |
federal false claims laws which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other government reimbursement programs that are false or fraudulent. The government may assert that a claim including items or services resulting from a violation of the federal healthcare program anti-kickback law or related to off-label promotion constitutes a false or fraudulent claim for purposes of the federal false claims laws; |
|
|
· |
the federal Health Insurance Portability and Accountability Act of 1996 which prohibits executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters and which also imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information; |
|
|
· |
the federal transparency requirements under the Health Care Reform Law requires manufacturers of drugs, devices, biologics, and medical supplies to report to the Department of Health and Human Services information related to physician payments and other transfers of value and physician ownership and investment interests; |
|
|
· |
the Federal Physician Payments Sunshine Act within the ACA, and its implementing regulations, require that certain manufacturers of drugs, devices, biological and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with certain exceptions) to report information related to certain payments or other transfers of value made or distributed to physicians and teaching hospitals, or to entities or individuals at the request of, or designated on behalf of, the physicians and teaching hospitals and to report annually certain ownership and investment interests held by physicians and their immediate family members; and |
|
|
· |
state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws, which may apply to items or services reimbursed by any third party payer, including commercial insurers, and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by federal laws, thus complicating compliance efforts. |
In addition, we may be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. The Health Insurance Portability and Accountability Act, or HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and its implementing regulations, imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s privacy and security standards directly applicable to “business associates”—independent contractors or agents of covered entities that receive or obtain protected health information in connection with providing a service on behalf of a covered entity. HITECH also created four new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts.
Post-Marketing Regulations
Following approval of a new product, a pharmaceutical company and the approved product are subject to continuing regulation by the FDA and other federal and state regulatory authorities, including, among other things, monitoring and recordkeeping activities, reporting to applicable regulatory authorities of adverse experiences with the product, providing the regulatory authorities with updated safety and efficacy information, product sampling and distribution requirements, and complying with promotion and advertising requirements, which include, among others, standards for direct-to-consumer advertising, restrictions on promoting drugs for uses or in patient populations not described in the drug’s approved labeling (known as “off-label use”), limitations on industry-sponsored scientific and educational activities, and requirements for promotional activities involving the internet. Although physicians may prescribe legally available drugs for off-label uses, manufacturers may not market or promote such off-label uses. Modifications or enhancements to the products or labeling or changes of site of manufacture are often subject to the approval of the FDA and other regulators, which may or may not be received or may result in a lengthy review process.
21
Employees
We have 12 full time employees. All of our employees are engaged in administration, finance, clinical, manufacturing, regulatory and business development functions. We believe our relations with our employees are good. We anticipate that the number of employees will grow as we continue to develop our product candidates. In addition, we utilize and will continue to utilize consultants, clinical research organizations and third parties to perform our pre-clinical studies, clinical studies, manufacturing and regulatory functions.
Emerging Growth Company
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. We will remain an emerging growth company until the earlier of (1) January 1, 2020, (2) the last day of the first fiscal year in which our annual gross revenues exceed $1 billion, (3) the date on which we become a “large accelerated filer” as defined in Rule 12b-2 under the Securities Exchange Act of 1934, as amended, which would occur if the market value of our common stock that is held by non-affiliates exceeds $700 million as of the last business day of our most recently completed second fiscal quarter or (4) the date on which we have issued more than $1 billion in non-convertible debt during the preceding three-year period.
For as long as we remain an “emerging growth company,” we may take advantage of certain exemptions from various reporting requirements that are applicable to public companies that are not “emerging growth companies” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation and financial statements in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote to approve executive compensation and shareholder approval of any golden parachute payments not previously approved. We are choosing to “opt out” of the extended transition periods available under the JOBS Act for complying with new or revised accounting standards, and intend to take advantage of the other reporting exemptions until we are no longer an “emerging growth company.”
Corporate Information
Corbus Pharmaceuticals, Inc. (formerly known as JB Therapeutics Inc.), was incorporated on April 24, 2009 under the laws of the State of Delaware. On April 11, 2014, JB Therapeutics, Inc. completed a merger with Corbus Pharmaceuticals Holdings, Inc. and changed its name to Corbus Pharmaceuticals, Inc. Upon the consummation of the merger, Corbus Pharmaceuticals, Inc. became a wholly owned subsidiary of Corbus Pharmaceuticals Holdings, Inc. which continues to operate the business of Corbus Pharmaceuticals, Inc. Our principal executive offices are located at 100 River Ridge Drive, Norwood, Massachusetts 02062, and our telephone number is (619) 963-0100. Our website address is www.corbuspharma.com.
We make available free of charge on or through the Investor Relations link on our website, www.corbuspharma.com, access to press releases and investor presentations, as well as all materials that we file electronically with the SEC, including our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports, filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act as soon as reasonably practicable after electronically filing such materials with, or furnishing them to, the SEC. During the period covered by this Form 10-K, we made all such materials available through our website as soon as reasonably practicable after filing such materials with the SEC. You may also read and copy any materials filed by us with the SEC at the SEC’s Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549, and you may obtain information on the operation of the Public Reference Room by calling the SEC in the United States at 1-800-SEC-0330. In addition, the SEC maintains an Internet website, www.sec.gov, that contains reports, proxy and information statements and other information that we file electronically with the SEC.
22
An investment in our common stock is speculative and illiquid and involves a high degree of risk including the risk of a loss of your entire investment. You should carefully consider the risks and uncertainties described below and the other information contained in this report and our other reports filed with the Securities and Exchange Commission. The risks set forth below are not the only ones facing us. Additional risks and uncertainties may exist that could also adversely affect our business, operations and financial condition. If any of the following risks actually materialize, our business, financial condition and/or operations could suffer. In such event, the value of our common stock could decline, and you could lose all or a substantial portion of the money that you pay for our common stock.
Risk Related to our Company and our Business
Risks Related to Our Financial Position and Need for Capital
We are a clinical stage pharmaceutical company with a limited operating history.
We are a clinical stage pharmaceutical company with a limited operating history. We must obtain FDA clearance of our Investigational New Drug applications, or INDs, before clinical trials can commence, and must receive regulatory approval of our New Drug Applications, or NDAs, before commercial sales of a product can commence. The likelihood of success of our business plan must be considered in light of the problems, substantial expenses, difficulties, complications and delays frequently encountered in connection with developing and expanding early-stage businesses and the regulatory and competitive environment in which we operate. Pharmaceutical product development is a highly speculative undertaking, involves a substantial degree of risk and is a capital-intensive business.
Accordingly, you should consider our prospects in light of the costs, uncertainties, delays and difficulties frequently encountered by companies in the early stages of development, especially clinical pharmaceutical companies such as ours. Potential investors should carefully consider the risks and uncertainties that a company with a limited operating history will face. In particular, potential investors should consider that we cannot assure you that we will be able to:
|
|
· |
receive FDA approval of INDs for commencing our clinical trials; |
|
|
· |
successfully implement or execute our current business plan, or that our business plan is sound; |
|
|
· |
Successfully manufacture our clinical product and establish commercial drug supply; |
|
|
· |
obtain DEA licenses necessary for the manufacturing of Resunab and for evaluating Resunab in our clinical trials; |
|
|
· |
successfully complete clinical trials and obtain regulatory approval for the marketing of Resunab; |
|
|
· |
secure market exclusivity and/or adequate intellectual property protection for Resunab; |
|
|
· |
attract and retain an experienced management and advisory team; |
|
|
· |
secure acceptance of Resunab in the medical community and with third party payors and consumers; |
|
|
· |
launch commercial sales of Resunab, whether alone or in collaboration with others; and |
|
|
· |
raise sufficient funds in the capital markets to effectuate our business plan including clinical development, regulatory approval and commercialization for Resunab. |
If we cannot successfully execute any one of the foregoing, our business may not succeed and your investment will be adversely affected.
We have incurred operating losses in each year since our inception and expect to continue to incur substantial losses for the foreseeable future. We may never become profitable or, if achieved, be able to sustain profitability.
We expect to incur substantial expenses without corresponding revenues unless and until we are able to obtain regulatory approval and successfully commercialize Resunab. We have been engaged in developing Resunab since 2009. To date, we have not generated any revenue from Resunab and we expect to incur significant expense to complete our clinical program for Resunab in the United States and elsewhere. We may never be able to obtain regulatory approval for the marketing of Resunab in any indication in the United States or internationally. Even if we are able to commercialize Resunab or any other product candidate, there can be no assurance that we will generate significant revenues or ever achieve profitability. Our net losses for the years ended December 31, 2015 and December 31, 2014 were approximately $8,851,000 and $2,540,000, respectively. As of December 31, 2015, we had an accumulated deficit of approximately $13,278,000.
23
If we were to obtain FDA approval for Resunab, we would expect that our research and development expenses will continue to increase as we advance to clinical trials for indications for the treatment of cystic fibrosis, systemic sclerosis and dermatomyositis. We may elect to pursue FDA approval for Resunab in other indications, which will result in significant additional research and development expenses. As a result, we expect to continue to incur substantial losses for the foreseeable future, and these losses will increase. We are uncertain when or if we will be able to achieve or sustain profitability. If we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods. Failure to become and remain profitable would impair our ability to sustain operations and adversely affect the price of our common stock and our ability to raise capital.
There is substantial doubt about our ability to continue as a going concern, which will affect our ability to obtain future financing and may require us to curtail our operations.
Our financial statements as of December 31, 2015 were prepared under the assumption that we will continue as a going concern. The independent registered public accounting firm that audited our 2015 financial statements, in their report, included an explanatory paragraph referring to our recurring losses since inception and expressing substantial doubt in our ability to continue as a going concern. Our financial statements do not include any adjustments that might result from the outcome of this uncertainty. At December 31, 2015, we had cash and cash equivalents of approximately $12.3 million. Our ability to continue as a going concern depends on our ability to obtain additional equity or debt financing, attain further operating efficiencies, reduce expenditures, and, ultimately, to generate revenue. We cannot assure you, however, that we will be able to achieve any of the foregoing.
Our cash or cash equivalents will only fund our operations for a limited time and we will need to raise additional capital to support our development and commercialization efforts.
We are currently operating at a loss and expect our operating costs will increase significantly as we incur costs related to the clinical trials for Resunab. As of December 31, 2015, our consolidated cash balance was approximately $12.3 million. We believe we have sufficient financial resources to fund our operations into at least the fourth quarter of 2016. As a result, our independent registered public accounting firm has expressed substantial doubt about our ability to continue as a going concern in their report on our financial statements.
We do not currently have any arrangements or credit facilities in place as a source of funds, and there can be no assurance that we will be able to raise sufficient additional capital on acceptable terms, or at all, and if we are not successful in raising additional capital, we may not be able to continue as a going concern. We may seek additional capital through a combination of private and public equity offerings, debt financings and strategic collaborations. Debt financing, if obtained, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, and could increase our expenses and require that our assets secure such debt.
Equity financing, if obtained, could result in dilution to our then existing stockholders and/or require such stockholders to waive certain rights and preferences. If such financing is not available on satisfactory terms, or is not available at all, we may be required to delay, scale back or eliminate the development of business opportunities and our operations and financial condition may be materially adversely affected. We can provide no assurances that any additional sources of financing will be available to us on favorable terms, if at all. In addition, if we are unable to secure sufficient capital to fund our operations, we might have to enter into strategic collaborations that could require us to share commercial rights to Resunab with third parties in ways that we currently do not intend or on terms that may not be favorable to us. If we choose to pursue additional indications and/or geographies for Resunab or otherwise expand more rapidly than we presently anticipate we may also need to raise additional capital sooner than expected.
Risks Related to Product Development, Regulatory Approval, Manufacturing and Commercialization
We depend entirely on the success of Resunab, which has not yet demonstrated efficacy in Phase 2 clinical trials. If we are unable to generate revenues from Resunab, our ability to create stockholder value will be limited.
Our only product candidate currently is Resunab, which has successfully completed Phase 1 safety studies and is being evaluated in Phase 2 clinical studies for cystic fibrosis, systemic sclerosis and dermatomyositis. We do not generate revenues from any FDA approved drug products and have no other product candidates in development. There is no guarantee that our Phase 2 clinical trials will be successful or that we will continue with clinical studies to support an approval from the FDA for any indication. We note that most drug candidates never reach the clinical development stage and even those that do reach clinical development have only a small chance of successfully completing clinical development and gaining regulatory approval. Therefore, our business currently depends entirely on the successful development, regulatory approval and commercialization of Resunab, which may never occur.
24
If we are not able to obtain any required regulatory approvals for Resunab, we will not be able to commercialize our only product candidate and our ability to generate revenue will be limited.
We must successfully complete clinical trials for Resunab before we can apply for marketing approval. Even if we complete our clinical trials, it does not assure FDA approval. Our Phase 2 clinical trials may be unsuccessful, which would materially harm our business. Even if these Phase 2 clinical trials are successful, we are required to conduct additional clinical trials to establish Resunab’s safety and efficacy, before a New Drug Application, or NDA, can be filed with the FDA for marketing approval of Resunab.
Clinical testing is expensive, is difficult to design and implement, can take many years to complete and is uncertain as to outcome. Success in early phases of pre-clinical and clinical trials does not ensure that later clinical trials will be successful, and interim results of a clinical trial do not necessarily predict final results. A failure of one or more of our clinical trials can occur at any stage of testing. We may experience numerous unforeseen events during, or as a result of, the clinical trial process that could delay or prevent our ability to receive regulatory approval or commercialize Resunab. The research, testing, manufacturing, labeling, packaging, storage, approval, sale, marketing, advertising and promotion, pricing, export, import and distribution of drug products are subject to extensive regulation by the FDA and other regulatory authorities in the United States and other countries, which regulations differ from country to country. We are not permitted to market Resunab as a prescription pharmaceutical product in the United States until we receive approval of an NDA from the FDA, or in any foreign countries until we receive the requisite approval from such countries. In the United States, the FDA generally requires the completion of clinical trials of each drug to establish its safety and efficacy and extensive pharmaceutical development to ensure its quality before an NDA is approved. Regulatory authorities in other jurisdictions impose similar requirements. Of the large number of drugs in development, only a small percentage result in the submission of an NDA to the FDA and even fewer are eventually approved for commercialization. We have never submitted an NDA to the FDA or comparable applications to other regulatory authorities. If our development efforts for Resunab, including regulatory approval, are not successful for its planned indications, or if adequate demand for Resunab is not generated, our business will be harmed.
Our success depends on the receipt of regulatory approval and the issuance of such regulatory approvals is uncertain and subject to a number of risks, including the following:
|
|
· |
the FDA or comparable foreign regulatory authorities or institutional review boards, or IRBs, may disagree with the design or implementation of our clinical trials; |
|
|
· |
we may not be able to provide acceptable evidence of Resunab’s safety and efficacy; |
|
|
· |
the results of our clinical trials may not be satisfactory or may not meet the level of statistical or clinical significance required by the FDA, European Medicines Agency, or EMA, or other comparable foreign regulatory authorities for marketing approval; |
|
|
· |
the dosing of Resunab in a particular clinical trial may not be at an optimal level; |
|
|
· |
patients in our clinical trials may suffer adverse effects for reasons that may or may not be related to Resunab; |
|
|
· |
the data collected from clinical trials may not be sufficient to support the submission of an NDA or other submission or to obtain regulatory approval in the United States or elsewhere; |
|
|
· |
the FDA or comparable foreign regulatory authorities may fail to approve the manufacturing processes or facilities of third- party manufacturers with which we contract for clinical and commercial supplies; and |
|
|
· |
the approval policies or regulations of the FDA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval. |
Failure to obtain regulatory approval for Resunab for the foregoing or any other reasons will prevent us from commercializing this product candidate as a prescription product, and our ability to generate revenue will be materially impaired. We cannot guarantee that regulators will agree with our assessment of the results of the clinical trials we intend to conduct in the future or that such trials will be successful. The FDA, EMA and other regulators have substantial discretion in the approval process and may refuse to accept any application or may decide that our data is insufficient for approval and require additional clinical trials, or pre-clinical or other studies. In addition, varying interpretations of the data obtained from pre-clinical and clinical testing could delay, limit or prevent regulatory approval of a product candidate.
25
We are a clinical stage company and we have not submitted an NDA or received regulatory approval to market Resunab in any jurisdiction. We have only limited experience in filing the applications necessary to gain regulatory approvals and expect to rely on consultants and third party contract research organizations, or CROs, with expertise in this area to assist us in this process. Securing FDA approval requires the submission of pre-clinical, clinical, and/or pharmacokinetic data, information about product manufacturing processes and inspection of facilities and supporting information to the FDA for each therapeutic indication to establish a product candidate’s safety and efficacy for each indication. Resunab may prove to have undesirable or unintended side effects, toxicities or other characteristics that may preclude our obtaining regulatory approval or prevent or limit commercial use with respect to one or all intended indications.
The process of obtaining regulatory approvals is expensive, often takes many years, if approval is obtained at all, and can vary substantially based upon, among other things, the type, complexity and novelty of the product candidates involved, the jurisdiction in which regulatory approval is sought and the substantial discretion of the regulatory authorities. Changes in the regulatory approval policy during the development period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for a submitted product application may cause delays in the approval or rejection of an application. Regulatory approval obtained in one jurisdiction does not necessarily mean that a product candidate will receive regulatory approval in all jurisdictions in which we may seek approval, but the failure to obtain approval in one jurisdiction may negatively impact our ability to seek approval in a different jurisdiction. Failure to obtain regulatory marketing approval for Resunab in any indication will prevent us from commercializing the product candidate, and our ability to generate revenue will be materially impaired.
Resunab is our only product candidate in development. If we fail to successfully commercialize Resunab, we may need to acquire additional product candidates and our business will be adversely affected.
We have never commercialized any product candidates and do not have any other compounds in pre-clinical testing, lead optimization or lead identification stages beyond Resunab. We cannot be certain that Resunab will prove to be sufficiently effective and safe to meet applicable regulatory standards for any indication. If we fail to successfully commercialize Resunab as a treatment for cystic fibrosis, systemic sclerosis, dermatomyositis or any other indication, whether as a stand-alone therapy or in combination with other treatments, our business would be adversely affected.
Even if we receive regulatory approval for Resunab, we still may not be able to successfully commercialize this product, and the revenue that we generate from its sales, if any, may be limited.
If approved for marketing, the commercial success of Resunab will depend upon its acceptance by the medical community, including physicians, patients and health care payors. The degree of market acceptance of Resunab will depend on a number of factors, including:
|
|
· |
demonstration of clinical safety and efficacy; |
|
|
· |
relative convenience, pill burden and ease of administration; |
|
|
· |
the prevalence and severity of any adverse effects; |
|
|
· |
the willingness of physicians to prescribe Resunab and of the target patient population to try new therapies; |
|
|
· |
efficacy of Resunab compared to competing products; |
|
|
· |
the introduction of any new products that may in the future become available to treat indications for which Resunab may be approved; |
|
|
· |
new procedures or methods of treatment that may reduce the incidences of any of the indications in which Resunab may show utility; |
|
|
· |
pricing and cost-effectiveness; |
|
|
· |
the inclusion or omission of Resunab in applicable treatment guidelines; |
|
|
· |
the effectiveness of our or any future collaborators’ sales and marketing strategies; |
|
|
· |
limitations or warnings contained in FDA-approved labeling; |
|
|
· |
our ability to obtain and maintain sufficient third-party coverage or reimbursement from government health care programs, including Medicare and Medicaid, private health insurers and other third-party payors; and |
|
|
· |
the willingness of patients to pay out-of-pocket in the absence of third-party coverage or reimbursement. |
26
If Resunab is approved, but does not achieve an adequate level of acceptance by physicians, health care payors and patients, we may not generate sufficient revenue and we may not be able to achieve or sustain profitability. Our efforts to educate the medical community and third-party payors on the benefits of Resunab may require significant resources and may never be successful.
In addition, even if we obtain regulatory approvals, the timing or scope of any approvals may prohibit or reduce our ability to commercialize Resunab successfully. For example, if the approval process takes too long, we may miss market opportunities and give other companies the ability to develop competing products or establish market dominance. Any regulatory approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render Resunab not commercially viable. For example, regulatory authorities may approve Resunab for fewer or more limited indications than we request, may not approve the price we intend to charge for Resunab, may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve Resunab with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that indication. Further, the FDA or comparable foreign regulatory authorities may place conditions on approvals, such as risk management plans and a Risk Evaluation and Mitigation Strategy, or REMS, to assure the safe use of the drug. If the FDA concludes a REMS is needed, the sponsor of the NDA must submit a proposed REMS; the FDA will not approve the NDA without an approved REMS, if required. A REMS could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA may also require a REMS for an approved product when new safety information emerges. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of Resunab. Moreover, product approvals may be withdrawn for non-compliance with regulatory standards or if problems occur following the initial marketing of the product. Any of the foregoing scenarios could materially harm the commercial success of Resunab.
Even if we obtain marketing approval for Resunab, we will be subject to ongoing obligations and continued regulatory review, which may result in significant additional expense. Additionally, Resunab could be subject to labeling and other restrictions and withdrawal from the market and we may be subject to penalties if we fail to comply with regulatory requirements or if we experience unanticipated problems with Resunab.
Even if we obtain United States regulatory approval of Resunab for an indication, the FDA may still impose significant restrictions on its indicated uses or marketing or the conditions of approval, or impose ongoing requirements for potentially costly and time-consuming post-approval studies, including Phase 4 clinical trials, and post-market surveillance to monitor safety and efficacy. Resunab will also be subject to ongoing regulatory requirements governing the manufacturing, labeling, packaging, storage, distribution, safety surveillance, advertising, promotion, recordkeeping and reporting of adverse events and other post-market information. These requirements include registration with the FDA, as well as continued compliance with current Good Clinical Practices regulations, or cGCPs, for any clinical trials that we conduct post-approval. In addition, manufacturers of drug products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with current Good Manufacturing Practices, or cGMP, requirements relating to quality control, quality assurance and corresponding maintenance of records and documents.
The FDA has the authority to require a risk evaluation and mitigation strategy, or REMS, as part of an NDA or after approval, which may impose further requirements or restrictions on the distribution or use of an approved drug, such as limiting prescribing to certain physicians or medical centers that have undergone specialized training, limiting treatment to patients who meet certain safe-use criteria or requiring patient testing, monitoring and/or enrollment in a registry.
With respect to sales and marketing activities by us or any future partner, advertising and promotional materials must comply with FDA rules in addition to other applicable federal, state and local laws in the United States and similar legal requirements in other countries. In the United States, the distribution of product samples to physicians must comply with the requirements of the U.S. Prescription Drug Marketing Act. Application holders must obtain FDA approval for product and manufacturing changes, depending on the nature of the change. We may also be subject, directly or indirectly through our customers and partners, to various fraud and abuse laws, including, without limitation, the U.S. Anti-Kickback Statute, U.S. False Claims Act, and similar state laws, which impact, among other things, our proposed sales, marketing, and scientific/educational grant programs. If we participate in the U.S. Medicaid Drug Rebate Program, the Federal Supply Schedule of the U.S. Department of Veterans Affairs, or other government drug programs, we will be subject to complex laws and regulations regarding reporting and payment obligations. All of these activities are also potentially subject to U.S. federal and state consumer protection and unfair competition laws. Similar requirements exist in many of these areas in other countries.
In addition, if Resunab is approved for an indication, our product labeling, advertising and promotion would be subject to regulatory requirements and continuing regulatory review. The FDA strictly regulates the promotional claims that may be made about prescription products. In particular, a product may not be promoted for uses that are not approved by the FDA as reflected in the product’s approved labeling. If we receive marketing approval for Resunab, physicians may nevertheless legally prescribe our products to their patients in a manner that is inconsistent with the approved label. If we are found to have promoted such off-label uses, we may become subject to significant liability and government fines. The FDA and other agencies actively enforce the laws and
27
regulations prohibiting the promotion of off-label uses by a company, and any company that is found to have improperly promoted off-label uses may be subject to significant sanctions. The federal government has levied large civil and criminal fines against companies for alleged improper promotion and has enjoined several companies from engaging in off-label promotion. The FDA has also requested that companies enter into consent decrees of permanent injunctions under which specified promotional conduct is changed or curtailed.
If we or a regulatory agency discover previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, problems with the facility where the product is manufactured, or if we or our manufacturers fail to comply with applicable regulatory requirements, we may be subject to the following administrative or judicial sanctions:
|
|
· |
restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, or voluntary or mandatory product recalls; |
|
|
· |
issuance of warning letters or untitled letters; |
|
|
· |
clinical holds; |
|
|
· |
injunctions or the imposition of civil or criminal penalties or monetary fines; |
|
|
· |
suspension of any ongoing clinical trials; |
|
|
· |
refusal to approve pending applications or supplements to approved applications filed by us, or suspension or revocation of product license approvals; |
|
|
· |
suspension or imposition of restrictions on operations, including costly new manufacturing requirements; or |
|
|
· |
product seizure or detention or refusal to permit the import or export of product. |
The occurrence of any event or penalty described above may inhibit our ability to commercialize Resunab and generate revenue. Adverse regulatory action, whether pre-or post-approval, can also potentially lead to product liability claims and increase our product liability exposure.
We currently have no sales and marketing organization. If we are unable to secure a sales and marketing partner or establish satisfactory sales and marketing capabilities, we may not successfully commercialize Resunab.
At present, we have no sales or marketing personnel. In order to commercialize products that are approved for commercial sales, we must either collaborate with third parties that have such commercial infrastructure or develop our own sales and marketing infrastructure. If we are not successful entering into appropriate collaboration arrangements, or recruiting sales and marketing personnel or in building a sales and marketing infrastructure, we will have difficulty successfully commercializing Resunab, which would adversely affect our business, operating results and financial condition.
We may not be able to enter into collaboration agreements on terms acceptable to us or at all. In addition, even if we enter into such relationships, we may have limited or no control over the sales, marketing and distribution activities of these third parties. Our future revenues may depend heavily on the success of the efforts of these third parties. If we elect to establish a sales and marketing infrastructure we may not realize a positive return on this investment. In addition, we will have to compete with established and well-funded pharmaceutical and biotechnology companies to recruit, hire, train and retain sales and marketing personnel. Factors that may inhibit our efforts to commercialize Resunab without strategic partners or licensees include:
|
|
· |
our inability to recruit and retain adequate numbers of effective sales and marketing personnel; |
|
|
· |
the inability of sales personnel to obtain access to or persuade adequate numbers of physicians to prescribe Resunab; |
|
|
· |
the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines; and |
|
|
· |
unforeseen costs and expenses associated with creating an independent sales and marketing organization. |
We face competition from other biotechnology and pharmaceutical companies and our operating results will suffer if we fail to compete effectively.
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. We have competitors in a number of jurisdictions, many of which have substantially greater name recognition, commercial infrastructures and financial, technical and personnel resources than we have. Established competitors may invest heavily to quickly discover and develop novel compounds that could make Resunab obsolete or uneconomical. Any new product that competes with an approved product may need to demonstrate compelling advantages in efficacy, cost, convenience, tolerability and safety to be
28
commercially successful. Other competitive factors, including generic competition, could force us to lower prices or could result in reduced sales. In addition, new products developed by others could emerge as competitors to Resunab. If we are not able to compete effectively against our current and future competitors, our business will not grow and our financial condition and operations will suffer.
Our potential competitors both in the United States and Europe include companies developing and/or marketing drugs for cystic fibrosis, including Vertex, Nivalis Therapeutics, ProQr, Proteostasis and PTC Therapeutics, as well as companies working in the systemic sclerosis field, including Bristol-Myers Squibb, Roche, Boehringer Ingelheim and Sanofi.
Recently enacted and future legislation may increase the difficulty and cost for us to obtain marketing approval of and commercialize Resunab and affect the prices we may obtain.
In the United States and some foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval for Resunab, restrict or regulate post-approval activities and affect our ability to profitably sell Resunab. Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. We do not know whether additional legislative changes will be enacted, or whether the FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals of Resunab, if any, may be. In addition, increased scrutiny by the U.S. Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
In the United States, the Medicare Modernization Act, or MMA, changed the way Medicare covers and pays for pharmaceutical products. The legislation expanded Medicare coverage for drug purchases by the elderly and introduced a new reimbursement methodology based on average sales prices for drugs. In addition, this legislation authorized Medicare Part D prescription drug plans to use formularies where they can limit the number of drugs that will be covered in any therapeutic class. As a result of this legislation and the expansion of federal coverage of drug products, we expect that there will be additional pressure to contain and reduce costs. These cost reduction initiatives and other provisions of this legislation could decrease the coverage and price that we receive for Resunab and could seriously harm our business. While the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates, and any reduction in reimbursement that results from the MMA may result in a similar reduction in payments from private payors.
In March 2010, President Obama signed into law the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act of 2010 or, collectively, the Health Care Reform Law, a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. Effective October 1, 2010, the Health Care Reform Law revised the definition of “average manufacturer price” for reporting purposes, which could increase the amount of Medicaid drug rebates to states. Further, the new law imposed a significant annual fee on companies that manufacture or import branded prescription drug products. Substantial new provisions affecting compliance have also been enacted, which may require us to modify our business practices with healthcare practitioners, and incur substantial costs to ensure compliance.
Despite initiatives to invalidate the Health Care Reform Law, at this time it appears the implementation of the Health Care Reform Law will continue. We will not know the full effects of the Health Care Reform Law until applicable federal and state agencies issue regulations or guidance under the new law. Although it is too early to determine the effect of the Health Care Reform Law, the new law appears likely to continue the pressure on pharmaceutical pricing, especially under the Medicare program, and may also increase our regulatory burdens and operating costs.
In addition, other legislative changes have been proposed and adopted in the United States since the Health Care Reform Law was enacted. On August 2, 2011, the Budget Control Act of 2011 among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, starting in 2013. On January 2, 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, or the ATRA, which delayed for another two months the budget cuts mandated by these sequestration provisions of the Budget Control Act of 2011. The ATRA, among other things, also reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. We expect that additional federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, and in turn could significantly reduce the projected value of certain development projects and reduce our profitability.
29
Our future growth depends, in part, on our ability to penetrate foreign markets, where we would be subject to additional regulatory burdens and other risks and uncertainties.
Our future profitability will depend, in part, on our ability to commercialize Resunab in foreign markets for which we intend to rely on collaborations with third parties. If we commercialize Resunab in foreign markets, we would be subject to additional risks and uncertainties, including:
|
|
· |
our customers’ ability to obtain reimbursement for Resunab in foreign markets; |
|
|
· |
our inability to directly control commercial activities because we are relying on third parties; |
|
|
· |
the burden of complying with complex and changing foreign regulatory, tax, accounting and legal requirements; |
|
|
· |
different medical practices and customs in foreign countries affecting acceptance in the marketplace; |
|
|
· |
import or export licensing requirements; |
|
|
· |
longer accounts receivable collection times; |
|
|
· |
longer lead times for shipping; |
|
|
· |
language barriers for technical training; |
|
|
· |
reduced protection of intellectual property rights in some foreign countries; |
|
|
· |
foreign currency exchange rate fluctuations; and |
|
|
· |
the interpretation of contractual provisions governed by foreign laws in the event of a contract dispute. |
Foreign sales of Resunab could also be adversely affected by the imposition of governmental controls, political and economic instability, trade restrictions and changes in tariffs, any of which may adversely affect our results of operations.
If we market Resunab in a manner that violates healthcare fraud and abuse laws, or if we violate government price reporting laws, we may be subject to civil or criminal penalties.
The FDA enforces laws and regulations which require that the promotion of pharmaceutical products be consistent with the approved prescribing information. While physicians may prescribe an approved product for a so-called “off label” use, it is unlawful for a pharmaceutical company to promote its products in a manner that is inconsistent with its approved label and any company which engages in such conduct may be subject to significant liability. Similarly, industry codes in the European Union and other foreign jurisdictions prohibit companies from engaging in off-label promotion and regulatory agencies in various countries enforce violations of the code with civil penalties. While we intend to ensure that our promotional materials are consistent with our label, regulatory agencies may disagree with our assessment and may issue untitled letters, warning letters or may institute other civil or criminal enforcement proceedings. In addition to FDA restrictions on marketing of pharmaceutical products, several other types of state and federal healthcare fraud and abuse laws have been applied in recent years to restrict certain marketing practices in the pharmaceutical industry. These laws include the U.S. Anti-Kickback Statute, U.S. False Claims Act and similar state laws. Because of the breadth of these laws and the narrowness of the safe harbors, it is possible that some of our business activities could be subject to challenge under one or more of these laws.
The U.S. Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce, or in return for, purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted broadly to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. Although there are several statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchasing or recommending may be subject to scrutiny if they do not qualify for an exemption or safe harbor. Our practices may not, in all cases, meet all of the criteria for safe harbor protection from anti-kickback liability. Moreover, recent health care reform legislation has strengthened these laws. For example, the Health Care Reform Law, among other things, amends the intent requirement of the U.S. Anti-Kickback Statute and criminal health care fraud statutes; a person or entity no longer needs to have actual knowledge of this statute or specific intent to violate it. In addition, the Health Care Reform Law provides that the government may assert that a claim including items or services resulting from a violation of the U.S. Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the U.S. False Claims Act. Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government or knowingly making, or causing to be made, a false statement to get a false claim paid.
30
Over the past few years, pharmaceutical and other healthcare companies have been prosecuted under these laws for a variety of alleged promotional and marketing activities, such as: allegedly providing free trips, free goods, sham consulting fees and grants and other monetary benefits to prescribers; reporting to pricing services inflated average wholesale prices that were then used by federal programs to set reimbursement rates; engaging in off-label promotion that caused claims to be submitted to Medicare or Medicaid for non-covered, off-label uses; and submitting inflated best price information to the Medicaid Rebate Program to reduce liability for Medicaid rebates. Most states also have statutes or regulations similar to the U.S. Anti-Kickback Statute and the U.S. False Claims Act, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor. Sanctions under these federal and state laws may include substantial civil monetary penalties, exclusion of a manufacturer’s products from reimbursement under government programs, substantial criminal fines and imprisonment.
We are, and will be, completely dependent on third parties to manufacture Resunab, and our commercialization of Resunab could be halted, delayed or made less profitable if those third parties fail to obtain manufacturing approval from the FDA or comparable foreign regulatory authorities, fail to provide us with sufficient quantities of Resunab or fail to do so at acceptable quality levels or prices.
We do not currently have, nor do we plan to acquire, the capability or infrastructure to manufacture the active pharmaceutical ingredient, or API, in Resunab for use in our clinical trials or for commercial product, if any. In addition, we do not have the capability to encapsulate Resunab as a finished drug product for commercial distribution. As a result, we will be obligated to rely on contract manufacturers, if and when Resunab is approved for commercialization. We have not entered into an agreement with any contract manufacturers for commercial supply and may not be able to engage a contract manufacturer for commercial supply of Resunab on favorable terms to us, or at all.
The facilities used by our contract manufacturers to manufacture Resunab must be approved by the FDA pursuant to inspections that will be conducted after we submit our NDA to the FDA. We do not control the manufacturing process of, and are completely dependent on, our contract manufacturing partners for compliance with cGMPs for manufacture of both active drug substances and finished drug products. These cGMP regulations cover all aspects of the manufacturing, testing, quality control and record keeping relating to Resunab. If our contract manufacturers cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA or others, they will not be able to secure and/or maintain regulatory approval for their manufacturing facilities. If the FDA or a comparable foreign regulatory authority does not approve these facilities for the manufacture of Resunab or if it withdraws any such approval in the future, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval for or market Resunab, if approved.
Our contract manufacturers will be subject to ongoing periodic unannounced inspections by the FDA and corresponding state and foreign agencies for compliance with cGMPs and similar regulatory requirements. We will not have control over our contract manufacturers’ compliance with these regulations and standards. Failure by any of our contract manufacturers to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, failure to grant approval to market Resunab, delays, suspensions or withdrawals of approvals, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect our business. In addition, we will not have control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and qualified personnel. Failure by our contract manufacturers to comply with or maintain any of these standards could adversely affect our ability to develop, obtain regulatory approval for or market Resunab.
If for any reason, these third parties are unable or unwilling to perform, we may not be able to terminate our agreements with them, and we may not be able to locate alternative manufacturers or formulators or enter into favorable agreements with them and we cannot be certain that any such third parties will have the manufacturing capacity to meet future requirements. If these manufacturers or any alternate manufacturer of finished drug product experiences any significant difficulties in its respective manufacturing processes for our API or finished Resunab product or should cease doing business with us, we could experience significant interruptions in the supply of Resunab or may not be able to create a supply of Resunab at all. Were we to encounter manufacturing issues, our ability to produce a sufficient supply of Resunab might be negatively affected. Our inability to coordinate the efforts of our third party manufacturing partners, or the lack of capacity available at our third party manufacturing partners, could impair our ability to supply Resunab at required levels. Because of the significant regulatory requirements that we would need to satisfy in order to qualify a new bulk or finished product manufacturer, if we face these or other difficulties with our current manufacturing partners, we could experience significant interruptions in the supply of Resunab if we decided to transfer the manufacture of Resunab to one or more alternative manufacturers in an effort to deal with the difficulties.
Any manufacturing problem or the loss of a contract manufacturer could be disruptive to our operations and result in lost sales. Additionally, we rely on third parties to supply the raw materials needed to manufacture our potential products. Any reliance on suppliers may involve several risks, including a potential inability to obtain critical materials and reduced control over production costs, delivery schedules, reliability and quality. Any unanticipated disruption to a future contract manufacturer caused by problems at suppliers could delay shipment of Resunab, increase our cost of goods sold and result in lost sales.
31
We cannot guarantee that our manufacturing and supply partners will be able to reduce the costs of commercial scale manufacturing of Resunab over time. If the commercial-scale manufacturing costs of Resunab are higher than expected, these costs may significantly impact our operating results. In order to reduce costs, we may need to develop and implement process improvements. However, in order to do so, we will need, from time to time, to notify or make submissions with regulatory authorities, and the improvements may be subject to approval by such regulatory authorities. We cannot be sure that we will receive these necessary approvals or that these approvals will be granted in a timely fashion. We also cannot guarantee that we will be able to enhance and optimize output in our commercial manufacturing process. If we cannot enhance and optimize output, we may not be able to reduce our costs over time.
Our product candidate aljuemic acid, Resunab, is currently classified as a Schedule I controlled substance subject to U.S. controlled substance laws and regulations, including regulations of the Drug Enforcement Agency and the U.S. Food and Drug Administration. Failure to obtain the necessary licenses and registrations and failure to comply with these laws could result in the delay in the manufacturing and distribution of Resunab and could delay the completion of clinical studies. Such delays and the cost of compliance with these laws and regulations, could adversely affect our business operations and our financial condition.
In the United States, our product candidate, Resunab, is currently classified as Schedule I controlled substance as defined in the Controller Substance Act (“CSA”). This designation is based on Resunab’s chemical structure and pharmacology (namely, it being a synthetic endocannabinoid mimetic that binds to the CB2 receptor). Even though Resunab’s mechanism of action is to modulate the immune system and results to date from clinical studies have demonstrated the drug has no psychotropic effects (which we believe is unlike other members of its chemical class), the DEA classifies Resunab as a Schedule I substance.
Schedule I controlled substances are pharmaceutical products subject to specific regulations under the CSA, that establishes, among other things, certain registration, manufacturing quotas, security, recordkeeping, reporting, import, export and other requirements administered by the DEA. All parties responsible for the manufacturing, distribution and testing the drug in clinical studies must apply for and obtain a license from the DEA before they are permitted to perform these activities with Resunab. Furthermore, these parties must have the security, control, recordkeeping, reporting and inventory mechanisms required by the DEA to prevent drug loss and diversion. All licensed facilities are required to renew their registrations annually if they intend to continue to work with our drug. The DEA conducts periodic inspections of certain registered establishments that handle controlled substances. We have been working with our manufacturers, distributors, exporters and clinical sites to obtain the necessary licenses to work with Resunab. The parties responsible for the manufacturing, distribution and export of Resunab have already applied for and have been granted DEA licenses and a number of institutions responsible for conducting our Phase 2 clinical studies have also been granted DEA licenses. However the failure to maintain the necessary registrations and the delay or failure of additional clinical sites to obtain DEA registrations, could delay the manufacturing, distribution and export of Resunab and could delay the completion of the Phase 2 clinical studies. Furthermore, failure to maintain compliance with the CSA, particularly non-compliance resulting in loss or diversion, could result in regulatory action that could have a material adverse effect on our business, financial condition and results of operations. The DEA may seek civil penalties, refuse to renew necessary registrations, or initiate proceedings to restrict, suspend or revoke those registrations. In certain circumstances, violations could lead to criminal proceedings. In addition, if the FDA, DEA, or any foreign regulatory authority determines that Resunab may have potential for abuse, it may require us to generate more clinical or other data than we currently anticipate to establish whether or to what extent the substance has an abuse potential, which could increase the cost and/or delay the launch of Resunab.
Individual states have also established controlled substance laws and regulations. Though state-controlled substances laws often mirror federal law, because the states are separate jurisdictions, they may separately schedule drugs, as well. While some states automatically schedule a drug based on federal action, other states schedule drugs through rulemaking or a legislative action. The requirement for state registrations could also result in delay of the manufacturing, distribution of Resunab or in the completion of the Phase 2 clinical studies. We and our manufacturing vendors and clinical sites must also obtain separate state registrations, permits or licenses in order to be able to obtain, handle, and distribute controlled substances for clinical trials or commercial sale, and failure to meet applicable regulatory requirements could lead to enforcement and sanctions by the states in addition to those from the DEA or otherwise arising under federal law.
The manufacturing and distribution of Resunab is subject to the DEA's annual manufacturing and procurement quota requirements. The annual quota allocated to us or our contract manufacturers for the controlled substances in Resunab may not be sufficient to complete clinical trials. Consequently, any delay or refusal by the DEA in establishing our, or our contract manufacturers, procurement and/or production quota for controlled substances could delay or stop our clinical trials or product launches, which could have a material adverse effect on our business, financial position and operations.
Delays in shipping Resunab could have a material adverse effect on our business, results of operations and financial condition.
The import and export of Resunab requires import and export licenses. However, because Resunab is currently a Schedule I controlled substance in the United States, in addition to the FDA and U.S. Customs and Border Protection, its import and export is
32
also regulated by the DEA. We may not be granted, or if granted, maintain, such licenses for import or export from the authorities these regulatory agencies. Even if we obtain the relevant licenses, shipments of Resunab may be held up in transit by any of these authorities, which could cause significant delays and may lead to product batches which no longer meet specifications for use in clinical trials or commercial distribution. Such events could result in delayed development timelines, increased expenses and partial or total loss of revenue from Resunab.
We expect that we will rely on third parties to conduct clinical trials for Resunab. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize Resunab and our business would be substantially harmed.
We expect to enter into agreements with third-party CROs to conduct and manage our clinical programs including contracting with clinical sites to perform our clinical studies. We plan to rely heavily on these parties for execution of clinical studies for Resunab and will control only certain aspects of their activities. Nevertheless, we will be responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards, and our reliance on CROs and clinical sites will not relieve us of our regulatory responsibilities. We and our CROs will be required to comply with cGCPs, which are regulations and guidelines enforced by the FDA, the Competent Authorities of the Member States of the European Economic Area and comparable foreign regulatory authorities for any products in clinical development. The FDA enforces these cGCP regulations through periodic inspections of trial sponsors, principal investigators and trial sites. If we or our CROs fail to comply with applicable cGCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that, upon inspection, the FDA will determine that any of our clinical trials comply with cGCPs. In addition, our clinical trials must be conducted with products produced under cGMP regulations and will require a large number of test subjects. Our failure or the failure of our CROs or clinical sites to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process and could also subject us to enforcement action up to and including civil and criminal penalties.
Although we intend to design the clinical trials for Resunab in consultation with CROs, we expect that the CROs will manage all of the clinical trials conducted at contracted clinical sites. As a result, many important aspects of our drug development programs would be outside of our direct control. In addition, the CROs and clinical sites may not perform all of their obligations under arrangements with us or in compliance with regulatory requirements. If the CROs or clinical sites do not perform clinical trials in a satisfactory manner, breach their obligations to us or fail to comply with regulatory requirements, the development and commercialization of Resunab for the subject indication may be delayed or our development program materially and irreversibly harmed. We cannot control the amount and timing of resources these CROs and clinical sites will devote to our program or Resunab. If we are unable to rely on clinical data collected by our CROs, we could be required to repeat, extend the duration of, or increase the size of our clinical trials, which could significantly delay commercialization and require significantly greater expenditures.
If any of our relationships with these third-party CROs or clinical sites terminate, we may not be able to enter into arrangements with alternative CROs or clinical sites. If CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other reasons, any such clinical trials may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for or successfully commercialize Resunab. As a result, our financial results and the commercial prospects for Resunab would be harmed, our costs could increase and our ability to generate revenue could be delayed.
Any termination or suspension of or delays in the commencement or completion of any necessary studies of Resunab for any indications could result in increased costs to us, delay or limit our ability to generate revenue and adversely affect our commercial prospects.
The commencement and completion of clinical studies can be delayed for a number of reasons, including delays related to:
|
|
· |
the FDA failing to grant permission to proceed and placing the clinical study on hold; |
|
|
· |
subjects failing to enroll or remain in our trials at the rate we expect; |
|
|
· |
a facility manufacturing Resunab being ordered by the FDA or other government or regulatory authorities to temporarily or permanently shut down due to violations of cGMP requirements or other applicable requirements, or cross-contaminations of product in the manufacturing process; |
|
|
· |
any changes to our manufacturing process that may be necessary or desired; |
|
|
· |
subjects choosing an alternative treatment for the indications for which we are developing Resunab, or participating in competing clinical studies; |
|
|
· |
subjects experiencing severe or unexpected drug-related adverse effects; |
33
|
|
· |
reports of similar technologies and products raising safety and/or efficacy concerns; |
|
|
· |
third-party clinical investigators losing their license or permits necessary to perform our clinical trials, not performing our clinical trials on our anticipated schedule or employing methods consistent with the clinical trial protocol, cGCP requirements, or other third parties not performing data collection and analysis in a timely or accurate manner; |
|
|
· |
inspections of clinical study sites by the FDA or IRBs finding regulatory violations that require us to undertake corrective action, result in suspension or termination of one or more sites or the imposition of a clinical hold on the entire study, or that prohibit us from using some or all of the data in support of our marketing applications; |
|
|
· |
third-party contractors becoming debarred or suspended or otherwise penalized by the FDA or other government or regulatory authorities for violations of regulatory requirements, in which case we may need to find a substitute contractor, and we may not be able to use some or any of the data produced by such contractors in support of our marketing applications; |
|
|
· |
one or more IRBs refusing to approve, suspending or terminating the study at an investigational site precluding enrollment of additional subjects, or withdrawing its approval of the trial; reaching agreement on acceptable terms with prospective CROs and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
|
|
· |
deviations of the clinical sites from trial protocols or dropping out of a trial; |
|
|
· |
adding new clinical trial sites; |
|
|
· |
the inability of the CRO to execute any clinical trials for any reason; and |
|
|
· |
government or regulatory delays or “clinical holds” requiring suspension or termination of a trial. |
Product development costs for Resunab will increase if we have delays in testing or approval or if we need to perform more or larger clinical studies than planned. Additionally, changes in regulatory requirements and policies may occur and we may need to amend study protocols to reflect these changes. Amendments may require us to resubmit our study protocols to the FDA and IRBs for reexamination, which may impact the costs, timing or successful completion of that study. If we experience delays in completion of, or if we, the FDA or other regulatory authorities, the IRB, or other reviewing entities, or any of our clinical study sites suspend or terminate any of our clinical studies of Resunab, its commercial prospects may be materially harmed and our ability to generate product revenues will be delayed. Any delays in completing our clinical trials will increase our costs, slow down our development and approval process and jeopardize our ability to commence product sales and generate revenues. Any of these occurrences may harm our business, financial condition and prospects significantly. In addition, many of the factors that cause, or lead to, termination or suspension of, or a delay in the commencement or completion of, clinical studies may also ultimately lead to the denial of regulatory approval of Resunab. In addition, if one or more clinical studies are delayed, our competitors may be able to bring products to market before we do, and the commercial viability of Resunab could be significantly reduced.
Clinical drug development involves a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results.
Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. The results of pre-clinical studies and early clinical trials may not be predictive of the results of later-stage clinical trials. We cannot assure you that the FDA will view the results as we do or that any future trials of Resunab will achieve positive results. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through pre-clinical studies and initial clinical trials. A number of companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier trials. Any future clinical trial results for Resunab may not be successful.
In addition, a number of factors could contribute to a lack of favorable safety and efficacy results for Resunab. For example, such trials could result in increased variability due to varying site characteristics, such as local standards of care, differences in evaluation period and surgical technique, and due to varying patient characteristics including demographic factors and health status.
34
We have been granted orphan drug designation for Resunab for the treatment of cystic fibrosis and systemic sclerosis. We also intend to seek orphan drug status for Resunab for the treatment of dermatomyositis. Upon receipt of regulatory approval, orphan drug status will provide us with seven years of market exclusivity in the United States under the Orphan Drug Act. However, there is no guarantee that the FDA will grant orphan drug designation for Resunab for dermatomyositis or any other indication, which would make us ineligible for the additional exclusivity and other benefits of orphan drug designation. Moreover, there can be no assurance that another company also holding orphan drug designation for the same indication or which may receive orphan drug designation in the future will not receive approval prior to us, in which our competitor would have the benefit of the seven year market exclusivity, and we would be unable to commercialize our product for the same indication until the expiration of the seven-year period. Even if we are the first to obtain approval for the orphan drug indication, there are circumstances under which a competing product may be approved for the same indication during our seven-year period of exclusivity.
Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making a drug available in the Unites States for this type of disease or condition will be recovered from sales of the product. Orphan drug designation must be requested before submitting an NDA. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan product designation does not convey any advantage in or shorten the duration of regulatory review and approval process. In addition to the potential period of exclusivity, orphan designation makes a company eligible for grant funding of up to $400,000 per year for four years to defray costs of clinical trial expenses, tax credits for clinical research expenses and potential exemption from the FDA application user fee.
If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan drug exclusivity, which means the FDA may not approve any other applications to market the same drug for the same indication for seven years, except in limited circumstances, such as (i) the drug’s orphan designation is revoked; (ii) its marketing approval is withdrawn; (iii) the orphan exclusivity holder consents to the approval of another applicant’s product; (iv) the orphan exclusivity holder is unable to assure the availability of a sufficient quantity of drug; or (v) a showing of clinical superiority to the product with orphan exclusivity by a competitor product. If a drug designated as an orphan product receives marketing approval for an indication broader than what is designated, it may not be entitled to orphan drug exclusivity. There can be no assurance that we will receive orphan drug designation for Resunab in the indications of cystic fibrosis, systemic sclerosis, or other inflammatory diseases, if we elect to seek such applications.
Third-party coverage and reimbursement and health care cost containment initiatives and treatment guidelines may constrain our future revenues.
Our ability to successfully market Resunab will depend in part on the level of reimbursement that government health administration authorities, private health coverage insurers and other organizations provide for the cost of our products and related treatments. Countries in which Resunab is expected to be sold through reimbursement schemes under national health insurance programs frequently require that manufacturers and sellers of pharmaceutical products obtain governmental approval of initial prices and any subsequent price increases. In certain countries, including the United States, government-funded and private medical care plans can exert significant indirect pressure on prices. We may not be able to sell Resunab profitably if adequate prices are not approved or coverage and reimbursement is unavailable or limited in scope. Increasingly, third-party payors attempt to contain health care costs in ways that are likely to impact our development of products including:
|
|
· |
failing to approve or challenging the prices charged for health care products; |
|
|
· |
introducing reimportation schemes from lower priced jurisdictions; |
|
|
· |
limiting both coverage and the amount of reimbursement for new therapeutic products; |
|
|
· |
denying or limiting coverage for products that are approved by the regulatory agencies but are considered to be experimental or investigational by third-party payors; and |
|
|
· |
refusing to provide coverage when an approved product is used in a way that has not received regulatory marketing approval. |
Risks Relating to Our Intellectual Property Rights
It is difficult and costly to protect our intellectual property rights, and we cannot ensure the protection of these rights.
Our commercial success will depend, in part, on obtaining and maintaining patent protection for our technologies, products and processes, successfully defending these patents against third-party challenges and successfully enforcing these patents against third party competitors. The patent positions of pharmaceutical companies can be highly uncertain and involve complex legal, scientific and factual questions for which important legal principles remain unresolved. Changes in either the patent laws or in interpretations of
35
patent laws may diminish the value of our intellectual property. Accordingly, we cannot predict the breadth of claims that may be allowable or enforceable in our patents (including patents owned by us). We currently have one issued patent and the three pending patent applications for Resunab may never be approved by United States or foreign patent offices and the existing patent and patent applications relating to Resunab and related technologies may be challenged, invalidated or circumvented by third parties and might not protect us against competitors with similar products or technologies.
The degree of future protection for our proprietary rights is uncertain, because legal means afford only limited protection and may not adequately protect our rights, permit us to gain or keep our competitive advantage, or provide us with any competitive advantage at all. For example, others have filed, and in the future are likely to file, patent applications covering products and technologies that are similar, identical or competitive to Resunab, or important to our business. We cannot be certain that any patent application owned by a third party will not have priority over patent applications filed by us, or that we will not be involved in interference, opposition or invalidity proceedings before United States or foreign patent offices.
We also rely on trade secrets to protect technology, especially in cases when we believe patent protection is not appropriate or obtainable. However, trade secrets are difficult to protect. While we require employees, academic collaborators, consultants and other contractors to enter into confidentiality agreements, we may not be able to adequately protect our trade secrets or other proprietary or licensed information. Typically, research collaborators and scientific advisors have rights to publish data and information in which we may have rights. If we cannot maintain the confidentiality of our proprietary technology and other confidential information, our ability to receive patent protection and our ability to protect valuable information owned by us may be imperiled. Enforcing a claim that a third-party entity illegally obtained and is using any of our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, courts are sometimes less willing to protect trade secrets than patents. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how.
If we fail to obtain or maintain patent protection or trade secret protection for Resunab or our technologies, third parties could use our proprietary information, which could impair our ability to compete in the market and adversely affect our ability to generate revenues and attain profitability.
We may also rely on the trademarks we may develop to distinguish our products from the products of our competitors. We cannot guarantee that any trademark applications filed by us or our business partners will be approved. Third parties may also oppose such trademark applications, or otherwise challenge our use of the trademarks. In the event that the trademarks we use are successfully challenged, we could be forced to rebrand our products, which could result in loss of brand recognition, and could require us to devote resources to advertising and marketing new brands. Further, we cannot provide assurance that competitors will not infringe the trademarks we use, or that we will have adequate resources to enforce these trademarks.
Resunab may infringe the intellectual property rights of others, which could increase our costs and delay or prevent our development and commercialization efforts.
Our success depends in part on avoiding infringement of the proprietary technologies of others. The pharmaceutical industry has been characterized by frequent litigation regarding patent and other intellectual property rights. Identification of third party patent rights that may be relevant to our proprietary technology is difficult because patent searching is imperfect due to differences in terminology among patents, incomplete databases and the difficulty in assessing the meaning of patent claims. Additionally, because patent applications are maintained in secrecy until the application is published, we may be unaware of third-party patents that may be infringed by commercialization of Resunab or any future product candidate. There may be certain issued patents and patent applications claiming subject matter that we may be required to license in order to research, develop or commercialize Resunab, and we do not know if such patents and patent applications would be available to license on commercially reasonable terms, or at all. Any claims of patent infringement asserted by third parties would be time-consuming and may:
|
|
· |
result in costly litigation; |
|
|
· |
divert the time and attention of our technical personnel and management; |
|
|
· |
prevent us from commercializing a product until the asserted patent expires or is held finally invalid or not infringed in a court of law; |
|
|
· |
require us to cease or modify our use of the technology and/or develop non-infringing technology; or |
|
|
· |
require us to enter into royalty or licensing agreements. |
Although no third party has asserted a claim of infringement against us, others may hold proprietary rights that could prevent Resunab from being marketed. Any patent-related legal action against us claiming damages and seeking to enjoin commercial activities relating to Resunab or our processes could subject us to potential liability for damages and require us to obtain a license to continue to manufacture or market Resunab or any future product candidates. We cannot predict whether we would prevail in any
36
such actions or that any license required under any of these patents would be made available on commercially acceptable terms, if at all. In addition, we cannot be sure that we could redesign Resunab or any future product candidates or processes to avoid infringement, if necessary. Accordingly, an adverse determination in a judicial or administrative proceeding, or the failure to obtain necessary licenses, could prevent us from developing and commercializing Resunab or a future product candidate, which could harm our business, financial condition and operating results.
A number of companies, including several major pharmaceutical companies, have conducted research on anti-inflammatory and anti-fibrosis therapies which resulted in the filing of many patent applications related to this research. If we were to challenge the validity of these or any issued United States patent in court, we would need to overcome a statutory presumption of validity that attaches to every issued United States patent. This means that, in order to prevail, we would have to present clear and convincing evidence as to the invalidity of the patent’s claims.
If we were to challenge the validity of these or any issued United States patent in an administrative trial before the Patent Trial and Appeal Board in the United States Patent and Trademark Office, we would have to prove that the claims are unpatentable by a preponderance of the evidence. There is no assurance that a jury and/or court would find in our favor on questions of infringement, validity or enforceability.
We may be subject to claims that we have wrongfully hired an employee from a competitor or that we or our employees have wrongfully used or disclosed alleged confidential information or trade secrets of their former employers.
As is commonplace in our industry, we employ individuals who were previously employed at other pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject in the future to claims that our employees or prospective employees are subject to a continuing obligation to their former employers (such as non-competition or non-solicitation obligations) or claims that our employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
General Company-Related Risks
We will need to grow the size of our organization, and we may experience difficulties in managing this growth.
We currently have twelve employees. As our development and commercialization plans and strategies develop, we will need to expand the size of our employee base for managerial, operational, sales, marketing, financial and other resources. Future growth would impose significant added responsibilities on members of management, including the need to identify, recruit, maintain, motivate and integrate additional employees. In addition, our management may have to divert a disproportionate amount of its attention away from our day-to-day activities and devote a substantial amount of time to managing these growth activities. Our future financial performance and our ability to commercialize Resunab and any other future product candidates and our ability to compete effectively will depend, in part, on our ability to effectively manage our future growth.
Future capital raises may dilute our existing stockholders’ ownership and/or have other adverse effects on our operations.
If we raise additional capital by issuing equity securities, our existing stockholders’ percentage ownership will be reduced and these stockholders may experience substantial dilution. We may also issue equity securities that provide for rights, preferences and privileges senior to those of our common stock. If we raise additional funds by issuing debt securities, these debt securities would have rights senior to those of our common stock and the terms of the debt securities issued could impose significant restrictions on our operations, including liens on our assets. If we raise additional funds through collaborations and licensing arrangements, we may be required to relinquish some rights to our technologies or candidate products, or to grant licenses on terms that are not favorable to us.
If we are not successful in attracting and retaining highly qualified personnel, we may not be able to successfully implement our business strategy. In addition, the loss of the services of certain key employees, including Yuval Cohen, our CEO, Mark Tepper, our President and Chief Scientific Officer, Barbara White, our Chief Medical Officer and Sean Moran, our Chief Financial Officer would adversely impact our business prospects.
Our ability to compete in the highly competitive pharmaceuticals industry depends in large part upon our ability to attract highly qualified managerial, scientific and medical personnel. In order to induce valuable employees to remain with us, we intend to provide employees with stock options that vest over time. The value to employees of stock options that vest over time will be significantly affected by movements in our stock price that we will not be able to control and may at any time be insufficient to counteract more lucrative offers from other companies.
37
Our management team has expertise in many different aspects of drug development and commercialization. However, we will need to hire additional personnel as we further develop Resunab. Competition for skilled personnel in our market is intense and competition for experienced scientists may limit our ability to hire and retain highly qualified personnel on acceptable terms. Despite our efforts to retain valuable employees, members of our management, scientific and medical teams may terminate their employment with us on short notice. In connection with the Merger, we entered into employment agreements with certain of our executive officers. However, these employment arrangements provide for at-will employment, which means that any of our employees could leave our employment at any time, with or without notice. The loss of the services of any of our executive officers or other key employees could potentially harm our business, operating results or financial condition. In particular, we believe that the loss of the services of Yuval Cohen Ph.D., our Chief Executive Officer, Mark Tepper Ph.D., our President and Chief Scientific Officer, Barbara White, M.D., our Chief Medical Officer and Sean Moran, C.P.A., M.B.A., our Chief Financial Officer, would have a material adverse effect on our business. Our success also depends on our ability to continue to attract, retain and motivate highly skilled junior, mid-level, and senior managers as well as junior, mid-level, and senior scientific and medical personnel.
Other pharmaceutical companies with which we compete for qualified personnel have greater financial and other resources, different risk profiles, and a longer history in the industry than we do. They also may provide more diverse opportunities and better chances for career advancement. Some of these characteristics may be more appealing to high-quality candidates than what we have to offer. If we are unable to continue to attract and retain high-quality personnel, the rate and success at which we can develop and commercialize product candidates would be limited.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of Resunab.
We face a potential risk of product liability as a result of the clinical testing of Resunab and will face an even greater risk if we commercialize Resunab or any other future product. For example, we may be sued if any product we develop, including Resunab, or any materials that we use in our products allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability and a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of Resunab. Even successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
|
|
· |
decreased demand for Resunab or any future products that we may develop; |
|
|
· |
injury to our reputation; |
|
|
· |
withdrawal of clinical trial participants; |
|
|
· |
costs to defend the related litigation; |
|
|
· |
a diversion of management’s time and our resources; |
|
|
· |
substantial monetary awards to trial participants or patients; |
|
|
· |
product recalls, withdrawals or labeling, marketing or promotional restrictions; |
|
|
· |
the inability to commercialize Resunab; and |
|
|
· |
a decline in the value of our stock. |
Our inability to obtain and retain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of products we develop. We intend to obtain product liability insurance covering our clinical trials. Although we will maintain such insurance, any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. Our insurance policies also have various exclusions, and we may be subject to a product liability claim for which we have no coverage. We may have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital to pay such amounts.
We may acquire businesses or products, or form strategic alliances, in the future, and we may not realize the benefits of such acquisitions.
We may acquire additional businesses or products, form strategic alliances or create joint ventures with third parties that we believe will complement or augment our existing business. If we acquire businesses with promising markets or technologies, we may not be able to realize the benefit of acquiring such businesses if we are unable to successfully integrate them with our existing operations and company culture. We may encounter numerous difficulties in developing, manufacturing and marketing any new
38
products resulting from a strategic alliance or acquisition that delay or prevent us from realizing their expected benefits or enhancing our business. We cannot assure you that, following any such acquisition, we will achieve the expected synergies to justify the transaction.
Risks Related to our Common Stock
Our affiliates may control our company for the foreseeable future, including the outcome of matters requiring stockholder approval.
Our officers, directors, and five percent stockholders collectively owned approximately 24.9% of our outstanding shares of common stock as of December 31, 2015. In addition, these stockholders entered into a voting agreement whereby they agreed to vote in favor of nominees for directors selected by the parties to the voting agreement as described herein. As a result, such entities and individuals may have the ability, acting together, to control the election of our directors and the outcome of corporate actions requiring stockholder approval, such as: (i) a merger or a sale of our company, (ii) a sale of all or substantially all of our assets, and (iii) amendments to our articles of incorporation and bylaws. This concentration of voting power and control could have a significant effect in delaying, deferring or preventing an action that might otherwise be beneficial to our other stockholders and be disadvantageous to our stockholders with interests different from those entities and individuals. Certain of these individuals also have significant control over our business, policies and affairs as officers or directors of our company. Therefore, you should not invest in reliance on your ability to have any control over our company.
An investment in our company should be considered illiquid.
An investment in our company requires a long-term commitment, with no certainty of return. Because we became a reporting company other than by the traditional means of conducting an initial public offering of our common stock, we may be unable to establish a liquid market for our common stock. In addition, investment banks may be less likely to agree to underwrite primary or secondary offerings on behalf of our company or its stockholders in the future than they would if we had become a public reporting company by means of an initial public offering of common stock. If all or any of the foregoing risks occur, it would have a material adverse effect on our company.
An active, liquid trading market for our common stock may not develop or be sustained.
Presently, our common stock is traded on the Nasdaq Capital Market and as we are in our early stages, an investment in our company will require a long-term commitment, with no certainty of return. Presently there is limited trading in our stock and in the absence of an active trading market:
|
|
· |
investors may have difficulty buying and selling or obtaining market quotations; |
|
|
· |
market visibility for shares of our common stock may be limited; and |
|
|
· |
a lack of visibility for shares of our common stock may have a depressive effect on the market price for shares of our common stock. |
The lack of an active market impairs your ability to sell your shares at the time you wish to sell them or at a price that you consider reasonable. The lack of an active market may also reduce the fair market value of your shares. An inactive market may also impair our ability to raise capital to continue to fund operations by selling shares and may impair our ability to acquire additional intellectual property assets by using our shares as consideration.
We are currently listed on the Nasdaq Capital Market. If we are unable to maintain listing of our securities on the Nasdaq Capital Market or any stock exchange, our stock price could be adversely affected and the liquidity of our stock and our ability to obtain financing could be impaired and it may be more difficult for our stockholders to sell their securities.
Although our common stock is currently listed on the Nasdaq Capital Market, we may not be able to continue to meet the exchange’s minimum listing requirements or those of any other national exchange. In addition, a liquid market may not develop for our common stock. If we are unable to maintain listing on the Nasdaq Capital Market or if a liquid market for our common stock does not develop, our common stock may remain thinly traded.
39
The Listing Rules of the Nasdaq Capital Market require listing issuers to comply with certain standards in order to remain listed on its exchange. If, for any reason, we should fail to maintain compliance with these listing standards and Nasdaq should delist our securities from trading on its exchange and we are unable to obtain listing on another national securities exchange, a reduction in some or all of the following may occur, each of which could have a material adverse effect on our stockholders:
|
|
· |
the liquidity of our common stock; |
|
|
· |
the market price of our common stock; |
|
|
· |
our ability to obtain financing for the continuation of our operations; |
|
|
· |
the number of institutional and general investors that will consider investing in our common stock; |
|
|
· |
the number of investors in general that will consider investing in our common stock; |
|
|
· |
the number of market makers in our common stock; |
|
|
· |
the availability of information concerning the trading prices and volume of our common stock; and |
|
|
· |
the number of broker-dealers willing to execute trades in shares of our common stock. |
Even if an active trading market for our common stock develops, the market price of our common stock may be significantly volatile.
Even if an active market for our common stock develops, of which no assurances can be given, the market price for our common stock may be volatile and subject to wide fluctuations in response to factors including the following:
|
|
· |
actual or anticipated fluctuations in our quarterly or annual operating results; |
|
|
· |
changes in financial or operational estimates or projections; |
|
|
· |
conditions in markets generally; |
|
|
· |
changes in the economic performance or market valuations of companies similar to ours; and |
|
|
· |
general economic or political conditions in the United States or elsewhere. |
In particular, the market prices of biotechnology companies like ours have been highly volatile due to factors, including, but not limited to:
|
|
· |
any delay or failure to conduct a clinical trial for our product or receive approval from the FDA and other regulatory agencies; |
|
|
· |
developments or disputes concerning our product’s intellectual property rights; |
|
|
· |
our or our competitors’ technological innovations; |
|
|
· |
changes in market valuations of similar companies; |
|
|
· |
announcements by us or our competitors of significant contracts, acquisitions, strategic partnerships, joint ventures, capital commitments, new technologies, or patents; and |
|
|
· |
failure to complete significant transactions or collaborate with vendors in manufacturing our product. |
The securities market has from time to time experienced significant price and volume fluctuations that are not related to the operating performance of particular companies. These market fluctuations may also materially and adversely affect the market price of shares of our common stock.
Future sales of shares by existing stockholders could cause our stock price to decline.
As of December 31, 2015, we had outstanding options to purchase an aggregate of 3,982,065 shares of our common stock at a weighted average exercise price of $1.03 per share and warrants to purchase an aggregate of 1,969,250 shares of our common stock at a weighted average exercise price of $0.97 per share. The exercise of such outstanding options and warrants will result in further dilution of your investment. If our existing stockholders sell substantial amounts of our common stock in the public market, or if the public perceives that such sales could occur, this could have an adverse impact on the market price of our common stock, even if there is no relationship between such sales and the performance of our business.
40
We are an “emerging growth company,” and will be able take advantage of reduced disclosure requirements applicable to “emerging growth companies,” which could make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or JOBS Act, and, for as long as we continue to be an “emerging growth company,” we intend to take advantage of certain exemptions from various reporting requirements applicable to other public companies but not to “emerging growth companies,” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. We will remain an emerging growth company until the earlier of (1) January 1, 2020, (2) the last day of the first fiscal year in which our annual gross revenues exceed $1 billion, (3) the date on which we become a “large accelerated filer” as defined in Rule 12b-2 under the Securities Exchange Act of 1934, as amended, which would occur if the market value of our common stock that is held by non-affiliates exceeds $700 million as of the last business day of our most recently completed second fiscal quarter or (4) the date on which we have issued more than $1 billion in non-convertible debt during the preceding three-year period.
We intend to take advantage of these reporting exemptions described above until we are no longer an “emerging growth company.” Under the JOBS Act, “emerging growth companies” can also delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not “emerging growth companies.”
We cannot predict if investors will find our common stock less attractive if we choose to rely on these exemptions. If some investors find our common stock less attractive as a result of any choices to reduce future disclosure, there may be a less active trading market for our common stock and our stock price may be more volatile.
We will incur significantly increased costs and devote substantial management time as a result of operating as a public company particularly after we are no longer an “emerging growth company.”
As a public company, we incur significant legal, accounting and other expenses that we did not incur as a private company. For example, we are required to comply with certain of the requirements of the Sarbanes-Oxley Act and the Dodd-Frank Wall Street Reform and Consumer Protection Act, as well as rules and regulations subsequently implemented by the SEC, including the establishment and maintenance of effective disclosure and financial controls and changes in corporate governance practices. We expect that compliance with these requirements will increase our legal and financial compliance costs and will make some activities more time consuming and costly. In addition, we expect that our management and other personnel will need to divert attention from operational and other business matters to devote substantial time to these public company requirements. In particular, we expect to incur significant expenses and devote substantial management effort toward ensuring compliance with the requirements of Section 404 of the Sarbanes-Oxley Act. In addition, after we are no longer qualify as an “emerging growth company,” we expect to incur additional management time and cost to comply with the more stringent reporting requirements applicable to companies that are deemed accelerated filers or large accelerated filers, including complying with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act. We currently do not have an internal audit function, and we will need to hire or contract for additional accounting and financial staff with appropriate public company experience and technical accounting knowledge.
We cannot predict or estimate the amount of additional costs we may incur as a result of becoming a public company or the timing of such costs.
There may be limitations on the effectiveness of our internal controls, and a failure of our control systems to prevent error or fraud may materially harm our company.
Proper systems of internal controls over financial accounting and disclosure are critical to the operation of a public company. As we are a start-up company, we have twelve full time employees which results in a lack of segregation of duties and are at the very early stages of establishing, and we may be unable to effectively establish such systems, especially in light of the fact that we expect to operate as a publicly reporting company. This would leave us without the ability to reliably assimilate and compile financial information about our company and significantly impair our ability to prevent error and detect fraud, all of which would have a negative impact on our company from many perspectives.
41
Moreover, we do not expect that disclosure controls or internal control over financial reporting, will prevent all error and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Further, the design of a control system must reflect the fact that there are resource constraints and the benefits of controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected. Failure of our control systems to prevent error or fraud could materially adversely impact us.
We do not currently intend to pay dividends on our common stock in the foreseeable future, and consequently, your ability to achieve a return on your investment will depend on appreciation in the price of our common stock.
We have never declared or paid cash dividends on our common stock and do not anticipate paying any cash dividends to holders of our common stock in the foreseeable future. Consequently, investors must rely on sales of their common stock after price appreciation, which may never occur, as the only way to realize any future gains on their investments. There is no guarantee that shares of our common stock will appreciate in value or even maintain the price at which our investors have purchased their shares.
We may be unable to complete our analysis of our internal controls over financial reporting in a timely manner, or these internal controls may not be determined to be effective, which may adversely affect investor confidence in our company and, as a result, the value of our common stock.
We are required, pursuant to Section 404 of the Sarbanes-Oxley Act, to furnish a report by our management on, among other things, the effectiveness of our internal control over financial reporting. This assessment will need to include disclosure of any material weaknesses identified by our management in our internal control over financial reporting.
We may not be able to complete our evaluation, testing and any required remediation in a timely fashion. During the evaluation and testing process, if we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal controls are effective.
If we are unable to assert that our internal control over financial reporting is effective, or, if applicable, our independent registered public accounting firm is unable to express an opinion on the effectiveness of our internal controls, we could lose investor confidence in the accuracy and completeness of our financial reports, which would cause the price of our common stock to decline, and we may be subject to investigation or sanctions by the SEC. We will also be required to disclose changes made in our internal control and procedures on a quarterly basis.
Our remediation efforts may not enable us to avoid a material weakness in our internal control over financial reporting in the future. Any of the foregoing occurrences, should they come to pass, could negatively impact the public perception of our company, which could have a negative impact on our stock price.
Upon dissolution of our company, you may not recoup all or any portion of your investment.
In the event of a liquidation, dissolution or winding-up of our company, whether voluntary or involuntary, the proceeds and/or assets of our company remaining after giving effect to such transaction, and the payment of all of our debts and liabilities and distributions required to be made to holders of any outstanding preferred stock will then be distributed to the stockholders of common stock on a pro rata basis. There can be no assurance that we will have available assets to pay to the holders of common stock, or any amounts, upon such a liquidation, dissolution or winding-up of our Company. In this event, you could lose some or all of your investment.
Our ability to use our net operating loss carryforwards and certain other tax attributes may be limited.
As a result of our merger in April 2014 with Corbus Pharmaceuticals, Inc., our wholly-owned subsidiary, our ability to utilize our federal net operating loss, carryforwards and federal tax credit may be limited under Sections 382 of the Internal Revenue Code. The limitations apply if an “ownership change,” as defined by Section 382, occurs. Generally, an ownership change occurs if the percentage of the value of the stock that is owned by one or more direct or indirect “five percent shareholders” increases by more than 50 percentage points over their lowest ownership percentage at any time during the applicable testing period (typically three years). In addition, future changes in our stock ownership, which may be outside of our control, may trigger an “ownership change” and, consequently, Section 382 limitations. As a result, if we earn net taxable income, our ability to use our pre-change net operating loss carryforwards and other tax attributes to offset United States federal taxable income may be subject to limitations, which could potentially result in increased future tax liability to us.
42
Our certificate of incorporation, as amended, allows for our board to create new series of preferred stock without further approval by our stockholders, which could adversely affect the rights of the holders of our common stock.
Our board of directors has the authority to fix and determine the relative rights and preferences of preferred stock. We anticipate that our board of directors will have the authority to issue up to 10,000,000 shares of our preferred stock without further stockholder approval. As a result, our board of directors could authorize the issuance of a series of preferred stock that would grant to holders the preferred right to our assets upon liquidation and the right to receive dividend payments before dividends are distributed to the holders of common stock. In addition, our board of directors could authorize the issuance of a series of preferred stock that has greater voting power than our common stock or that is convertible into our common stock, which could decrease the relative voting power of our common stock or result in dilution to our existing stockholders.
Not applicable.
Our principal offices are located at 100 River Ridge Drive, Norwood, MA 02062. We signed a three year lease for office space, commencing July 1, 2014, which included rent payments of approximately $168,000 in the aggregate. In August 2015, this lease was amended for our relocation into 6,326 square feet of new space within the existing building. In January 2016, we began occupying the office space under this lease amendment, which is for a five-year term and includes rent payments of approximately $872,000 in the aggregate. We believe our facilities are adequate for our foreseeable needs.
We are not currently subject to any material legal proceedings. However, we may from time to time become a party to various legal proceedings arising in the ordinary course of our business.
Not applicable.
43
PART II
|
Item 5. |
MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information
Our common stock is listed on the Nasdaq Capital Market under the symbol “CRBP.” Our shares of common stock began trading on the Nasdaq Capital Market under the symbol “CRBP” effective April 16, 2015. Prior to April 16, 2015, our common stock was quoted on the Over-the Counter Markets (the “OTC.QB”) under the symbol “CRBP.” Our shares of common stock began being quoted on the OTC.QB effective October 24, 2014.
The following table contains information about the range of high and low sale prices for our common stock for each quarter during the last two years. The source of these high and low sales prices was the Nasdaq Capital Market and the OTC.QB.
|
Fiscal Year Ended December 31, 2015 |
|
High Sales Price |
|
|
Low Sales Price |
|
||
|
First Quarter, |
|
$ |
3.25 |
|
|
$ |
2.00 |
|
|
Second Quarter |
|
$ |
4.31 |
|
|
$ |
2.63 |
|
|
Third Quarter |
|
$ |
4.22 |
|
|
$ |
1.45 |
|
|
Fourth Quarter |
|
$ |
2.55 |
|
|
$ |
1.50 |
|
|
Fiscal Year Ended December 31, 2014 |
|
High Sales Price |
|
|
Low Sales Price |
|
||
|
Fourth Quarter |
|
$ |
4.95 |
|
|
$ |
2.71 |
|
Dividends
We have never declared or paid cash dividends on our common stock. We do not intend to declare or pay cash dividends on our common stock for the foreseeable future, but currently intend to retain any future earnings to fund the development and growth of our business. The payment of cash dividends if any, on the common stock will rest solely within the discretion of our board of directors and will depend, among other things, upon our earnings, capital requirements, financial condition, and other relevant factors.
Record Holders
As of March 24, 2016, there are approximately 222 record holders of shares of common stock.
Not applicable.
44
The following discussion and analysis of our financial condition and results of operations should be read together with our financial statements and the related notes and the other financial information included elsewhere in this Annual Report. This discussion contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those anticipated in these forward-looking statements as a result of various factors, including those discussed below and elsewhere in this Annual Report, particularly those under “Risk Factors.”
Overview
We are a clinical stage pharmaceutical company, focused on the development and commercialization of novel therapeutics to treat rare or uncommon chronic and serious inflammatory and fibrotic diseases with clear unmet medical needs. Our product Resunab is a novel synthetic oral endocannabinoid-mimetic drug that is intended to resolve chronic inflammation and halt fibrotic processes without causing immunosuppression. Resunab is currently being evaluated in three separate Phase 2 studies for the treatment of cystic fibrosis, diffuse cutaneous systemic sclerosis and skin-predominant dermatomyositis. The cystic fibrosis and systemic sclerosis studies are expected to be completed by the end of 2016 and the dermatomyositis study is expected to be completed in the first half of 2017. The United States Food and Drug Administration has granted Resunab Orphan Drug Designation as well as Fast Track Status for both cystic fibrosis and systemic sclerosis. A fourth Phase 2 study of Resunab in systemic lupus erythematosus, or SLE is planned to start in the first quarter of 2017
Resunab is a synthetic, rationally-designed oral small molecule drug that selectively binds to the cannabinoid receptor type 2, or CB2, which is found on activated immune cells, fibroblasts and muscle cells. Resunab stimulates the production of Specialized Pro-Resolving Lipid Mediators, or SPMs, which act to resolve inflammation, clear bacteria and halt fibrosis by activating endogenous pathways. These endogenous resolution pathways are normally activated in healthy individuals during the course of normal immune responses but are dysfunctional in chronic inflammatory and fibrotic diseases. Through its’ activation of the CB2 receptor, Resunab is designed to move innate immune responses from the activation phase through completion of the resolution phase. The CB2 receptor plays an endogenous role in modulating and resolving inflammation by, in effect, turning heightened inflammation “off” and restoring homeostasis.
Financial Operations Overview
We are a research and development company and have not generated any revenues from the sale of products. We have never been profitable and, from inception through December 2015, our losses from operations have been approximately $13.3 million. Our net losses for the years ended December 31, 2015 and 2014 were approximately $8,851,000 and $2,540,000, respectively. We expect to continue to incur significant expenses and increasing operating losses for the foreseeable future. We expect our expenses to increase significantly in connection with our ongoing activities to develop, seek regulatory approval and commercialization of Resunab. Furthermore, we expect to incur additional costs associated with operating as a public company. Accordingly, we will need additional financing to support our continuing operations. We will seek to fund our operations through public or private equity or debt financings or other sources, which may include government grants and collaborations with third parties. Adequate additional financing may not be available to us on acceptable terms, or at all. Our failure to raise capital as and when needed would have a negative impact on our financial condition and our ability to pursue our business strategy. We will need to generate significant revenues to achieve profitability, and we may never do so.
We expect to continue to incur significant expenses and increasing operating losses for at least the next several years. We expect our expenses will increase substantially in 2016 and in the future in connection with our ongoing activities, as we:
|
|
· |
conduct clinical trials for Resunab in scleroderma, cystic fibrosis, systemic lupus erythematosus and other indications; |
|
|
· |
continue our research and development efforts; |
|
|
· |
manufacture clinical study materials and develop commercial scale manufacturing capabilities; |
|
|
· |
seek regulatory approval for our product candidates; |
|
|
· |
add personnel to support development of our product candidates; and |
|
|
· |
operate as a public company. |
45
Critical Accounting Policies and Estimates
Our consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America. The preparation of these financial statements requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period.
On an ongoing basis, we evaluate our estimates and judgments for all assets and liabilities, including those related to stock-based compensation expense and the fair value determined for stock purchase warrants classified as derivative liabilities. We base our estimates and judgments on historical experience, current economic and industry conditions and on various other factors that are believed to be reasonable under the circumstances. This forms the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
We believe that full consideration has been given to all relevant circumstances that we may be subject to, and the consolidated financial statements accurately reflect our best estimate of the results of operations, financial position and cash flows for the periods presented.
Revenue
To date, we have not generated any revenues from the sales of products. We do not expect to generate revenue from product sales unless and until we successfully complete development and obtain regulatory approval for the marketing of Resunab, which we expect will take a number of years and is subject to significant uncertainty.
To date, we have recorded $648,382 of collaboration revenue related to an award agreement we entered into in fiscal 2015 with the CFFT, a non-profit drug discovery and development affiliate of the Cystic Fibrosis Foundation, pursuant to which we received a development award (the “Award”) for up to $5 million in funding. The funding from the Award is supporting the Phase 2 clinical trial of Resunab in adults with cystic fibrosis. Upon the execution of the Award agreement, we received a payment of $1,250,000 in May 2015. In November 2015, we received a second payment of $1,250,000 upon the achievement of a milestone for dosing the first patient. We recorded these two milestone payments received from the CFFT totaling $2,500,000 as deferred revenue and they are being amortized on a straight-line basis over the expected duration of the performance period of the development program under the Award, which is expected to conclude in February 2017. The remaining $2,500,000 under the Award will be paid to us incrementally upon the achievement of certain milestones related to the progress of the Phase 2 CF clinical trial, as set forth in the Award agreement.
Research and Development
Research and development expenses are incurred for the development of Resunab and consist primarily of payroll, and payments to contract research and development companies. To date, these costs are related to generating pre-clinical data and the cost of manufacturing Resunab for clinical trials and conducting clinical trials. These costs are expected to increase significantly in the future as Resunab is evaluated in clinical trials.
General and Administrative Expenses
General and administrative expenses consist primarily of payroll, rent and professional services. Other general and administrative expenses include accounting and legal services. We anticipate that our general and administrative expenses will increase significantly during 2016 and in the future as we increase our headcount to support our continued research and development and the potential commercialization of our product candidates. We also anticipate increased expenses related to audit, legal, regulatory, and tax-related services associated with maintaining compliance with NASDAQ exchange listing and SEC requirements, director and officer insurance, and investor relations costs associated with being a public company.
Total Other Income (Expense)
Total other income (expense) consists primarily of interest income we earn on interest-bearing accounts, interest expense incurred on our outstanding debt, and gains or losses related to foreign currency exchange rate fluctuations. Additionally, in fiscal 2014 amounts included in other income (expense) included a gain on the settlement of debt, a gain related to the forgiveness of a note payable, and a loss related to the change in the fair value of a warrant liability.
46
Accrued Research and Development Expenses
As part of the process of preparing financial statements, we are required to estimate and accrue expenses, the largest of which are research and development expenses. This process involves: communicating with our applicable personnel to identify services that have been performed on our behalf and estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of actual cost; estimating and accruing expenses in our financial statements as of each balance sheet date based on facts and circumstances known to us at the time; and periodically confirming the accuracy of our estimates with selected service providers and making adjustments, if necessary.
Examples of estimated research and development expenses that we accrue include:
|
|
· |
fees paid to CROs in connection with nonclinical studies; |
|
|
· |
fees paid to contract manufacturers in connection with the production of Resunab for clinical trials ; |
|
|
· |
fees paid to CRO and research institutions in connection with conducting of clinical studies; and |
|
|
· |
professional service fees for consulting and related services. |
We base our expense accruals related to clinical studies on our estimates of the services performed pursuant to contracts with multiple research institutions and clinical research organizations that conduct and manage clinical studies on our behalf. The financial terms of these agreements vary from contract to contract and may result in uneven payment flows. Payments under some of these contracts depend on factors, such as the successful enrollment of patients and the completion of clinical study milestones. Our service providers invoice us monthly in arrears for services performed. In accruing service fees, we estimate the time period over which services will be performed and the level of effort to be expended in each period. If we do not identify costs that we have begun to incur or if we underestimate or overestimate the level of services performed or the costs of these services, our actual expenses could differ from our estimates.
To date, we have not experienced significant changes in our estimates of accrued research and development expenses following each applicable reporting period. However, due to the nature of estimates, we cannot assure you that we will not make changes to our estimates in the future as we become aware of additional information regarding the status or conduct of our clinical studies and other research activities.
Stock-Based Compensation
Stock options are granted with an exercise price at no less than fair market value at the date of the grant. The stock options normally expire ten years from the date of grant. Stock option awards vest upon terms determined by our board of directors.
We recognize compensation costs resulting from the issuance of stock-based awards to employees, members of our Board of directors and consultants. The fair value of each option grant was estimated as of the date of grant using the Black-Scholes option-pricing model. The fair value is amortized as compensation cost on a straight-line basis over the requisite service period of the awards, which is generally the vesting period. Due to our limited operating history and limited volume of sales of our common stock, we estimated our volatility in consideration of a number of factors, including the volatility of comparable public companies and, commencing in 2015, we also included the volatility of our own common stock. We use historical data, as well as subsequent events occurring prior to the issuance of the consolidated financial statements, to estimate option exercise and employee forfeitures within the valuation model. The expected term of options granted to employees under our stock plans is based on the average of the contractual term (generally 10 years) and the vesting period (generally 48 months). The expected term of options granted under the 2014 Plan, all of which qualify as “plain vanilla” per SEC Staff Accounting Bulletin 107, is based on the average of the 6.25 years. For non-employee options, the expected term is the contractual term and stock options granted to non-employee consultants are revalued at the end of each reporting period until vested and changes in their fair value are recorded as adjustments to expense over the related vesting period. The risk-free rate is based on the yield of a U.S. Treasury security with a term consistent with the option. The risk-free rate is based on the yield of a U.S. Treasury security with a term consistent with the expected term of the option. We estimate the forfeiture rate at the time of grant and revise it, if necessary, in subsequent periods if actual forfeitures differ from those estimates. Forfeitures were estimated based on management’s expectation through industry knowledge and historical data. We have never paid dividends on our common stock and do not anticipate paying dividends on our common stock in the foreseeable future. Accordingly, we have assumed no dividend yield for purposes of estimating the fair value of our share-based compensation.
47
The following assumptions were used to estimate the fair value of stock options granted using the Black-Scholes option pricing model for the years ended December 31, 2015 and 2014 is as follows:
|
|
|
2015 |
|
|
2014 |
|
||
|
Risk free interest rate |
|
|
1.85 |
% |
|
|
1.88 |
% |
|
Expected dividend yield |
|
|
0 |
% |
|
|
0 |
% |
|
Expected term in years |
|
|
6.73 |
|
|
|
6.20 |
|
|
Expected volatility |
|
|
90.68 |
% |
|
|
89.75 |
% |
|
Estimated forfeiture rate |
|
|
4.83 |
% |
|
|
19.20 |
% |
Valuation of Common Stock
Until October 24, 2014, when our common stock started trading on the Over-the-Counter Markets (“OTC.QB”), we were a privately held company with no active public market for our common stock. Our board of directors intends all options granted to be exercisable at a price per share not less than the per share fair value of our common stock underlying those options on the date of grant. The estimated fair value of our common stock was determined at each valuation date in accordance with the guidelines outlined in the American Institute of Certified Public Accountants Practice Aid, Valuation of Privately-Held-Company Equity Securities Issued as Compensation. Our board of directors, with the assistance of management and an outside third-party valuation firm, developed these valuations using significant judgment and taking into account numerous factors, including the stage of development of our company and market conditions.
For all options granted after the completion of the Series A financing in March 2012 until January 2014, the fair value of our common stock was supported by an independent third-party valuation. In conducting this valuation, we utilized the Market Approach to estimate the fair value of our common stock. Under this approach, we considered the market value of comparable early-stage public life science companies and used the ratio of market value to invested capital as a common benchmark for assessing valuation.
For the stock options granted on April 11, 2014 through October 2, 2014, we estimated the fair value of our stock to be $0.63 per share. This was based on our Private Placement completed in April and May 2014 (the “2014 Private Placement”), in which we sold one share of common stock and one warrant for $1.00. We estimated the fair value of each warrant to be $0.37 based on the Black Scholes valuation model. For stock options granted after October 2, 2014 but prior to October 24, 2014 we estimated the fair value of our stock to be $1.00 with the increase in value attributable to the effectiveness of a Registration Statement filed on Form S-1 (File No. 333-198563). Commencing on October 24, 2014, when our common stock started trading on the OTC.QB, we used the closing stock price on the date of grant as quoted on the OTC.QB to determine fair value. Commencing on April 16, 2015, when our common stock started trading on the Nasdaq Capital Market, we used the closing stock price on the date of grant as quoted on the Nasdaq Capital Market to determine fair value.
Emerging Growth Company Status
Under Section 107(b) of the Jumpstart Our Business Startups Act of 2012, emerging growth companies can delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not emerging growth companies.
Results of Operations
Comparison of Year Ended 2015 to 2014
Collaboration Revenue. On April 20, 2015, we entered into an award agreement with the CFFT, a non-profit drug discovery and development affiliate of the Cystic Fibrosis Foundation, pursuant to which we received a development award (the “Award”) for up to $5 million in funding. The funding from the Award is supporting the Phase 2 clinical trial of Resunab-inflammatory in adults with cystic fibrosis (“CF”). Upon the execution of the Award agreement, we received a payment of $1,250,000 in May 2015. In November 2015, we received$1,250,000 from the CFFT upon the achievement of a milestone for dosing the first patient. In fiscal 2015, we recorded these two milestone payments received from the CFFT totaling $2,500,000 as deferred revenue. We are amortizing the $2,500,000 on a straight-line basis over the expected duration of the performance period of the development program under the award, which is expected to conclude in February 2017. Accordingly, we recorded $648,382 of collaboration revenue in fiscal 2015. The remaining $2,500,000 that the Company can receive under the Award will be paid to the Company incrementally upon the achievement of certain milestones related to the progress of the Phase 2 CF clinical trial, as set forth in the Award agreement.
Research and Development. Research and Development expenses for the year ended December 31, 2015 totaled approximately $5,889,000, an increase of $4,633,000 over the $1,256,000 recorded for the year ended December 31, 2014. The increase in fiscal
48
2015 as compared to fiscal 2014 was primarily attributable to increases of approximately $3,769,000 for costs related to staffing, clinical operations and clinical trials as Resunab is currently being tested in three separate Phase 2 studies for the treatment of cystic fibrosis, diffuse cutaneous systemic sclerosis and skin-predominant dermatomyositis. Other increases in fiscal 2015 as compared to fiscal 2014 included $359,000 for the manufacturing of Resunab for clinical trials, $261,000 of regulatory costs, primarily for consulting services, $165,000 for stock-based compensation costs, and $79,000 for product development costs.
General and Administrative. General and Administrative expense for the year ended December 31, 2015 totaled approximately $3,613,000, an increase of $2,221,000 over the $1,392,000 recorded for year ended December 31, 2014. The increase in fiscal 2015 as compared to fiscal 2014 was primarily attributable to increases of approximately $795,000 in stock-based compensation costs, $649,000 in public company costs, $216,000 in insurance costs, $163,000 in compensation and recruiting costs, $157,000 in consulting costs and $97,000 in legal costs.
Other Income, Net. Other Income, Net for the year ended December 31, 2015 totaled approximately $3,000, a decrease of $104,000 compared to $107,000 for the year ended December 31, 2014. The decrease was primarily attributable to transactions in 2014 that did not recur in 2015 including a $145,000 gain on the settlement of debt and a $7,000 gain on the forgiveness of interest on a note payable, partially offset by a $28,000 charge related to the change in the fair value of a warrant liability.
Liquidity and Capital Resources
Since inception, we have experienced negative cash flows from operations. We have financed our operations primarily through sales of equity-related securities. In addition, the majority of the costs of the dermatomyositis and systemic lupus erythematosus clinical trials are being funded by NIH grants and our cystic fibrosis clinical trial is being partially funded by a $5 million award from the Cystic Fibrosis Foundation Trust. At December 31, 2015, our accumulated deficit since inception was approximately $13,278,000.
At December 31, 2015, we had total current assets of approximately $12,715,000 and current liabilities of approximately $3,630,000 resulting in working capital of $9,085,000. At December 31, 2015, we had total assets of approximately $12,875,000 and total liabilities of approximately $3,890,000, resulting in a stockholders’ equity of $8,985,000.
Net cash used in operating activities for the year ended December 31, 2015 was approximately $4,647,000, which includes a net loss of $8,851,000, non-cash expenses of $1,195,000, and $3,008,000 of cash provided from a change in net working capital items.
Cash used in investing activities for the year ended December 31, 2015 totaled approximately $114,000 for the purchase of property and equipment.
Cash provided from financing activities for the year ended December 31, 2015 totaled approximately $10,837,000 primarily from the issuance of common stock and warrants.
At December 31, 2015, we had total debt outstanding of approximately $162,000, which matures and is payable in the third quarter of 2016.
At December 31, 2015, we had a cash balance of approximately $12,338,000. We expect our current cash on hand to be sufficient to meet our operating and capital requirements into the fourth quarter of 2016. We will need to raise significant additional capital to continue to fund operations and the clinical trials for Resunab. We may seek to sell common or preferred equity or convertible debt securities, enter into a credit facility or another form of third-party funding, or seek other debt financing. In addition, we may seek to raise cash through collaborative agreements or from government grants. The sale of equity and convertible debt securities may result in dilution to our stockholders and those securities may have rights senior to those of our common shares. If we raise additional funds through the issuance of preferred stock, convertible debt securities or other debt financing, these securities or other debt could contain covenants that would restrict our operations. Any other third-party funding arrangement could require us to relinquish valuable rights.
The source, timing and availability of any future financing will depend principally upon market conditions, and, more specifically, on the progress of our clinical development programs. Funding may not be available when needed, at all, or on terms acceptable to us. Lack of necessary funds may require us, among other things, to delay, scale back or eliminate expenses including some or all of our planned clinical trials.
49
Contractual Obligations and Commitments
The following table presents information about our known contractual obligations as of December 31, 2015. It does not reflect contractual obligations that may have arisen or may arise after that date. Except for historical facts, the information in this section is forward‑looking information.
|
|
|
Payments due by period |
|
|||||||||||||||||
|
Contractual Obligations |
|
Total |
|
|
2016 |
|
|
Fiscal 2017-2018 |
|
|
Fiscal 2019-2020 |
|
|
After Fiscal 2020 |
|
|||||
|
Operating lease obligations (1) |
|
$ |
876,515 |
|
|
$ |
250,768 |
|
|
$ |
299,958 |
|
|
$ |
312,610 |
|
|
$ |
13,179 |
|
|
Capital lease obligations (2) |
|
|
12,711 |
|
|
|
3,884 |
|
|
|
8,474 |
|
|
353 |
|
|
— |
|
||
|
Total |
|
$ |
889,226 |
|
|
$ |
254,652 |
|
|
$ |
308,432 |
|
|
$ |
312,963 |
|
|
$ |
13,179 |
|
|
(1) |
In August 2015, we entered into an amendment to our office space lease agreement for our relocation into 6,326 square feet of new space within the existing building. In January 2016, we began occupying the office space under this lease amendment, which is for a five-year term and includes rent payments of approximately $872,000 in the aggregate. |
|
(2) |
On December 30, 2015, we entered into a lease agreement for a copier machine. The machine was placed in service in January 2016. The lease is for a three-year term and includes a bargain purchase option at the end of the term. |
We may enter into contracts in the normal course of business with clinical research organizations for clinical trials and clinical supply manufacturing and with vendors for pre-clinical research studies, research supplies and other services and products for operating purposes. These contracts generally provide for termination on notice, and therefore, we believe that our non-cancelable obligations under these agreements are not material. As of December 31, 2015, other than our lease for office space, we had no material Contractual Obligations or Commitments that will affect our future liquidity.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that is material to investors.
Not applicable.
See pages F-1 through F-20 following the Exhibit Index of this Annual Report on Form 10-K.
None.
Evaluation of Our Disclosure Controls
We maintain disclosure controls and procedures that are designed to provide reasonable assurance that material information required to be disclosed in our periodic reports filed under the Exchange Act is recorded, processed, summarized, and reported within the time periods specified in the SEC’s rules and forms and to provide reasonable assurance that such information is accumulated and communicated to our management, our chief executive officer and our chief financial officer, to allow timely decisions regarding required disclosure. We carried out an evaluation, under the supervision and with the participation of our management, including our principal executive and principal financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures, as defined in Rule 13(a)-15(e) under the Exchange Act. Based on this evaluation, our principal executive officer and principal financial officer concluded that, as of December 31, 2015, our disclosure controls and procedures were effective.
50
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on criteria established in the framework in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on this evaluation, our management concluded that our internal control over financial reporting was effective as of December 31, 2015.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risks that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
This Annual Report does not include an attestation report of our independent registered public accounting firm because we are not an accelerated filer or a large accelerated filer.
Changes in Internal Controls over Financial Reporting
In the fourth fiscal quarter of 2015, we hired a Controller in our finance department to enhance and strengthen our internal controls over financial reporting.
Other than the enhancement reported above, during the year ended December 31, 2015, there have been no changes in our internal control over financial reporting that have materially affected or are reasonably likely to materially affect our internal controls over financial reporting. From time to time, we make changes to our internal control over financial reporting that are intended to enhance its effectiveness and which do not have a material effect on our overall internal control over financial reporting.
None.
51
PART III
Information required by this item is incorporated by reference to our Proxy Statement for the 2016 Annual Meeting of Stockholders.
Information required by this item is incorporated herein by reference to our Proxy Statement for the 2016 Annual Meeting of Stockholders.
|
Item 12. |
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
Information required by this item is incorporated by reference to our Proxy Statement for the 2016 Annual Meeting of Stockholders.
Information required by this item is incorporated by reference to our Proxy Statement for the 2016 Annual Meeting of Stockholders.
Information required by this item is incorporated by reference to our Proxy Statement for the 2016 Annual Meeting of Stockholders.
52
PART IV
(a) List of Documents filed as part of this Report
(1) Consolidated Financial Statements
The financial statements and related notes, together with the report of EisnerAmper LLP appear at pages F-1 through F-19 following the Exhibit List as required by Part II, Item 8 “Financial Statements and Supplementary Data” of this Form 10-K.
(2) Financial Statement Schedules.
Schedules are omitted because they are either not required, not applicable, or the information is otherwise included.
(3) Exhibits
The Company has filed with this report or incorporated by reference herein certain exhibits as specified below pursuant to Rule 12b-32 under the Exchange Act. See Exhibit Index following the signature page to this report for a complete list of documents filed with this report.
|
Exhibit No. |
|
Description |
|
|
|
|
|
3.1 |
|
Certificate of Incorporation (incorporated by reference to Exhibit 3.1 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
3.2 |
|
Certificate of Amendment (incorporated by reference to Exhibit 3.2 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
3.3 |
|
Bylaws (incorporated by reference to Exhibit 3.3 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
4.1 |
|
Form of Merger Warrant (incorporated by reference to Exhibit 4.1 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
4.2 |
|
Form of Replacement Warrant (incorporated by reference to Exhibit 4.2 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
4.3 |
|
Form of Investor Warrant (incorporated by reference to Exhibit 4.3 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
4.4 |
|
Form of Additional Replacement Warrant (incorporated by reference to Exhibit 4.4 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
4.5 |
|
Form of Placement Agent Warrant (incorporated by reference to Exhibit 4.5 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
4.6 |
|
Registration Rights Agreement (incorporated by reference to Exhibit 4.6 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
4.7 |
|
Specimen Common Stock Certificate, $0.0001 par value (incorporated herein by reference to Exhibit 4.1 of the Company’s Registration Statement on Form S-3 filed with the SEC on November 10, 2015). |
|
|
|
|
|
5.1 |
|
Opinion of Lowenstein Sandler LLP (incorporated by reference to Exhibit 5.1 of the Company’s Registration Statement on Amendment No. 1 to Form S-1 filed with the SEC on September 30, 2014). |
|
|
|
|
|
10.1 |
|
Placement Agency Agreement, dated March 27, 2014, between the Company and Aegis Capital Corporation (incorporated by reference to Exhibit 10.1 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
10.2 |
|
Consulting Agreement, dated March 21, 2014, between the Company and Orchestra Medical Ventures (incorporated by reference to Exhibit 10.2 of the Company’s Registration Statement on Form S-1 filed with the SEC on |
53
|
Exhibit No. |
|
Description |
|
|
|
|
|
10.3 |
|
Form of Subscription Agreement for the Company’s 2014 Private Placement (incorporated by reference to Exhibit 10.3 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
10.4 |
|
Form of Voting Agreement, dated April 11, 2014, by and among the Company and the stockholders named therein (incorporated by reference to Exhibit 10.4 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
10.5 |
|
2014 Equity Compensation Plan (incorporated by reference to Exhibit 10.5 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
10.6 |
|
Form of Incentive Stock Option Agreement (incorporated by reference to Exhibit 10.6 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
10.7 |
|
Form of Non-Qualified Stock Option Agreement (incorporated by reference to Exhibit 10.7 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
10.8 |
|
Form of Restricted Stock Agreement (incorporated by reference to Exhibit 10.8 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
10.9 |
|
Employment Agreement, dated April 11, 2014, between the Company and Yuval Cohen (incorporated by reference to Exhibit 10.9 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
10.10 |
|
Employment Agreement, dated April 11, 2014, between the Company and Mark Tepper (incorporated by reference to Exhibit 10.10 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
10.11 |
|
Amended and Restated Employment Agreement, dated June 19, 2014, between the Company and Sean Moran (incorporated by reference to Exhibit 10.11 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
10.12 |
|
Agreement and Plan of Merger, dated March 27, 2014, by and among the Company, Corbus Pharmaceuticals Acquisition, Inc. and JB Therapeutics, Inc. (incorporated by reference to Exhibit 10.12 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
10.13 |
|
Subscription Agreement, dated April 2009, between Sumner Burstein and JB Therapeutics, Inc. (which is now known as Corbus Pharmaceuticals, Inc.) (incorporated by reference to Exhibit 10.13 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
10.14 |
|
Letter Agreement, dated April 29, 2009, between JB Therapeutics, Inc. (which is now known as Corbus Pharmaceuticals, Inc.) and Sumner Burstein (incorporated by reference to Exhibit 10.14 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
10.15 |
|
Form of Indemnification Agreement (incorporated by reference to Exhibit 10.15 of the Company’s Registration Statement on Amendment No. 1 to Form S-1 filed with the SEC on September 30, 2014). |
|
|
|
|
|
10.16 |
|
Letter Agreement, dated August 18, 2014, between the Company and Barbara White (incorporated herein by reference to Exhibit 10.15 of the Company’s Post-Effective Amendment No. 1 to Form S-1 filed with the SEC on |
|
|
|
|
|
10.17 |
|
Award Agreement, dated April 9, 2015, between Cystic Fibrosis Foundation Therapeutics, Inc. and the Company (incorporated herein by reference to Exhibit 10.1 of the Company’s Quarterly Report on Form 10-Q filed with the SEC on May 13, 2015).# |
|
|
|
|
|
21.1 |
|
List of Subsidiaries of the Company (incorporated by reference to Exhibit 21.1 of the Company’s Registration Statement on Form S-1 filed with the SEC on September 3, 2014). |
|
|
|
|
|
23.1 |
|
Consent of EisnerAmper LLP.* |
|
|
|
|
|
31.1 |
|
Certification of Chief Executive Officer pursuant to Rule 13a-14(a) or Rule 15d-14(a).* |
|
|
|
|
|
31.2 |
|
Certification of Chief Financial Officer pursuant to Rule 13a-14(a) or Rule 15d-14(a).* |
|
|
|
|
|
32.1 |
|
Certification of Chief Executive Officer pursuant to Rule 13a-14(b) or Rule 15d-14(b).* |
|
|
|
|
|
32.2 |
|
Certification of Chief Financial Officer pursuant to Rule 13a-14(b) or Rule 15d-14(b).* |
|
|
|
|
|
101.INS |
|
XBRL Instance Document.* |
|
|
|
|
54
|
Exhibit No. |
|
Description |
|
101.SCH |
|
XBRL Taxonomy Extension Schema Document.* |
|
|
|
|
|
101.CAL |
|
XBRL Taxonomy Extension Calculation Linkbase Document.* |
|
|
|
|
|
101.DEF |
|
XBRL Taxonomy Extension Definition Linkbase Document.* |
|
|
|
|
|
101.LAB |
|
XBRL Taxonomy Extension Label Linkbase Document.* |
|
|
|
|
|
101.PRE |
|
XBRL Taxonomy Extension Presentation Linkbase Document.* |
|
* |
Filed herewith. |
|
# |
Confidential treatment has been granted with respect to certain portions of this exhibit. Omitted portions have been submitted separately to the SEC. |
55
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
|
CORBUS PHARMACEUTICALS HOLDINGS, INC. |
||
|
|
|
|
|
|
|
Date: March 28, 2016 |
|
By: |
/s/ YUVAL COHEN |
|
|
|
|
|
Name: |
Yuval Cohen |
|
|
|
|
Title: |
Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
Signature |
|
Title |
|
Date |
|
|
|
|
|
|
|
/s/ YUVAL COHEN |
|
Chief Executive Officer and Director (Principal Executive Officer) |
|
March 28, 2016 |
|
Yuval Cohen |
|
|
||
|
|
|
|
|
|
|
/s/ SEAN MORAN |
|
Chief Financial Officer (Principal Financial and Accounting Officer) |
|
March 28, 2016 |
|
Sean Moran |
|
|
||
|
|
|
|
|
|
|
/s/ ALAN HOLMER |
|
Director |
|
March 28, 2016 |
|
Alan Holmer |
|
|
||
|
|
|
|
|
|
|
/s/ DAVID HOCHMAN |
|
Director |
|
March 28, 2016 |
|
David Hochman |
|
|
||
|
|
|
|
|
|
|
/s/ RENU GUPTA |
|
Director |
|
March 28, 2016 |
|
Renu Gupta |
|
|
||
|
|
|
|
|
|
|
/s/ AVERY CATLIN |
|
Director |
|
March 28, 2016 |
|
Avery Catlin |
|
|
56
INDEX TO FINANCIAL STATEMENTS
|
|
|
Page Number |
|
Corbus Pharmaceuticals Holdings, Inc. Financial Statements-December 31, 2015 |
|
|
|
|
F-2 |
|
|
Consolidated Balance Sheets as of December 31, 2015 and December 31, 2014 |
|
F-3 |
|
Consolidated Statements of Operations for the Years Ended December 31, 2015 and 2014 |
|
F-4 |
|
|
F-5 |
|
|
Consolidated Statements of Cash Flows for the Years Ended December 31, 2015 and 2014 |
|
F-6 |
|
|
F-7 |
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders
Corbus Pharmaceuticals Holdings, Inc.
We have audited the accompanying consolidated balance sheets of Corbus Pharmaceuticals Holdings, Inc. and Subsidiary (the “Company”) as of December 31, 2015 and 2014, and the related consolidated statements of operations, preferred stock and stockholders’ equity (deficit), and cash flows for each of the years then ended. The financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of Corbus Pharmaceuticals Holdings, Inc. and Subsidiary as of December 31, 2015 and 2014, and the consolidated results of their operations and their cash flows for each of the years then ended, in conformity with accounting principles generally accepted in the United States of America.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the consolidated financial statements, the Company has incurred recurring losses from operations which raises substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 2. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ EisnerAmper LLP
Iselin, New Jersey
March 28, 2016
F-2
Corbus Pharmaceuticals Holdings, Inc.
|
|
|
December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
ASSETS |
|
|
|
|
|
|
|
|
|
Current assets: |
|
|
|
|
|
|
|
|
|
Cash and cash equivalents |
|
$ |
12,338,275 |
|
|
$ |
6,262,445 |
|
|
Prepaid expenses |
|
|
376,515 |
|
|
|
270,556 |
|
|
Total current assets |
|
|
12,714,790 |
|
|
|
6,533,001 |
|
|
Restricted cash |
|
|
36,375 |
|
|
|
13,728 |
|
|
Property and equipment, net |
|
|
124,138 |
|
|
|
54,044 |
|
|
Total assets |
|
$ |
12,875,303 |
|
|
$ |
6,600,773 |
|
|
LIABILITIES AND STOCKHOLDERS’ EQUITY |
|
|
|
|
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
|
|
|
|
Notes payable |
|
$ |
162,019 |
|
|
$ |
144,389 |
|
|
Accounts payable |
|
|
1,314,377 |
|
|
|
344,160 |
|
|
Accrued expenses |
|
|
562,279 |
|
|
|
249,491 |
|
|
Deferred revenue, current |
|
|
1,591,358 |
|
|
|
— |
|
|
Total current liabilities |
|
|
3,630,033 |
|
|
|
738,040 |
|
|
Deferred revenue, noncurrent |
|
|
260,260 |
|
|
|
— |
|
|
Total liabilities |
|
|
3,890,293 |
|
|
|
738,040 |
|
|
Commitments and Contingencies |
|
|
|
|
|
|
|
|
|
Stockholders’ equity |
|
|
|
|
|
|
|
|
|
Preferred Stock $0.0001 par value:10,000,000 shares authorized, no shares issued and outstanding at December 31, 2015 and December 31, 2014 |
|
|
— |
|
|
|
— |
|
|
Common stock, $0.0001 par value; 150,000,000 shares authorized, 37,605,134 and 25,938,332 shares issued and outstanding at December 31, 2015 and December 31, 2014 |
|
|
3,761 |
|
|
|
2,594 |
|
|
Additional paid-in capital |
|
|
22,259,063 |
|
|
|
10,287,214 |
|
|
Accumulated deficit |
|
|
(13,277,814 |
) |
|
|
(4,427,075 |
) |
|
Total stockholders’ equity |
|
|
8,985,010 |
|
|
|
5,862,733 |
|
|
Total liabilities and stockholders’ equity |
|
$ |
12,875,303 |
|
|
$ |
6,600,773 |
|
The accompanying notes are an integral part of these Consolidated Financial Statements.
F-3
Corbus Pharmaceuticals Holdings, Inc.
Consolidated Statements of Operations
|
|
|
For the Years Ended December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
Collaboration revenue |
|
$ |
648,382 |
|
|
$ |
— |
|
|
Operating expenses: |
|
|
|
|
|
|
|
|
|
Research and development |
|
|
5,888,659 |
|
|
|
1,255,535 |
|
|
General and administrative |
|
|
3,613,416 |
|
|
|
1,391,638 |
|
|
Total operating expenses |
|
|
9,502,075 |
|
|
|
2,647,173 |
|
|
Operating loss |
|
|
(8,853,693 |
) |
|
|
(2,647,173 |
) |
|
Other income (expense): |
|
|
|
|
|
|
|
|
|
Interest expense |
|
|
(2,440 |
) |
|
|
(24,021 |
) |
|
Interest income |
|
|
3,417 |
|
|
|
2,115 |
|
|
Forgiveness of interest on note payable |
|
|
— |
|
|
|
7,466 |
|
|
Gain on the settlement of debt |
|
|
— |
|
|
|
145,006 |
|
|
Change in fair value of warrant liability |
|
|
— |
|
|
|
(28,448 |
) |
|
Foreign currency exchange gain |
|
|
1,977 |
|
|
|
4,570 |
|
|
Other income, net |
|
|
2,954 |
|
|
|
106,688 |
|
|
Net loss |
|
$ |
(8,850,739 |
) |
|
$ |
(2,540,485 |
) |
|
Net loss per share, basic and diluted |
|
$ |
(0.28 |
) |
|
$ |
(0.13 |
) |
|
Weighted average number of common shares outstanding, basic and diluted |
|
|
31,350,145 |
|
|
|
20,159,861 |
|
The accompanying notes are an integral part of these Consolidated Financial Statements.
F-4
Corbus Pharmaceuticals Holdings, Inc.
Statements of Preferred Stock and Stockholders’ Equity (Deficit)
|
|
|
Series A Preferred Stock Convertible |
|
|
Non-Convertible |
|
|
Common Stock |
|
|
Additional |
|
|
Accumulated |
|
|
Total Stockholders’ |
|
||||||||||||||||||
|
|
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
Shares |
|
|
Amount |
|
|
Paid-in Capital |
|
|
Deficit |
|
|
Equity (Deficit) |
|
|||||||||
|
Balance at December 31, 2013 |
|
|
1,835,212 |
|
|
$ |
1,106,609 |
|
|
|
200,000 |
|
|
$ |
2,000 |
|
|
|
6,964,788 |
|
|
$ |
696 |
|
|
$ |
102,696 |
|
|
$ |
(1,886,590 |
) |
|
$ |
(1,783,198 |
) |
|
Issuance of common stock at inception of Corbus |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
6,000,000 |
|
|
|
600 |
|
|
|
(600 |
) |
|
|
|
|
|
|
— |
|
|
Conversion of Series A convertible preferred stock into common stock upon reverse acquisition |
|
|
(1,835,212 |
) |
|
|
(1,106,609 |
) |
|
|
(200,000 |
) |
|
|
(2,000 |
) |
|
|
2,035,212 |
|
|
|
204 |
|
|
|
1,108,405 |
|
|
|
|
|
|
|
1,108,609 |
|
|
Issuance of common stock in private placement, net of issuance costs of $1,857,668 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10,260,000 |
|
|
|
1,026 |
|
|
|
8,401,306 |
|
|
|
|
|
|
|
8,402,332 |
|
|
Issuance of common stock in exchange for settlement of debt |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
541,948 |
|
|
|
54 |
|
|
|
396,138 |
|
|
|
|
|
|
|
396,192 |
|
|
Stock compensation expense |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
193,120 |
|
|
|
|
|
|
|
193,120 |
|
|
Issuance of common stock upon exercise of warrants |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
48,693 |
|
|
|
5 |
|
|
|
33,328 |
|
|
|
|
|
|
|
33,333 |
|
|
Issuance of common stock upon exercise of stock options |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
87,691 |
|
|
|
9 |
|
|
|
4,441 |
|
|
|
|
|
|
|
4,450 |
|
|
Reclassification of derivative warrant liability |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
48,380 |
|
|
|
|
|
|
|
48,380 |
|
|
Net Loss |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(2,540,485 |
) |
|
|
(2,540,485 |
) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at December 31, 2014 |
|
|
— |
|
|
$ |
— |
|
|
|
— |
|
|
$ |
— |
|
|
|
25,938,332 |
|
|
$ |
2,594 |
|
|
$ |
10,287,214 |
|
|
$ |
(4,427,075 |
) |
|
$ |
5,862,733 |
|
|
Stock compensation expense |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1,153,302 |
|
|
|
|
|
|
|
1,153,302 |
|
|
Issuance of common stock upon exercise of warrants, net of issuance costs of $509,215 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
11,615,674 |
|
|
|
1,162 |
|
|
|
10,812,963 |
|
|
|
|
|
|
|
10,814,125 |
|
|
Issuance of common stock upon exercise of stock options |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
51,128 |
|
|
|
5 |
|
|
|
5,584 |
|
|
|
|
|
|
|
5,589 |
|
|
Net Loss |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(8,850,739 |
) |
|
|
(8,850,739 |
) |
|
Balance at December 31, 2015 |
|
|
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
|
37,605,134 |
|
|
$ |
3,761 |
|
|
$ |
22,259,063 |
|
|
$ |
(13,277,814 |
) |
|
$ |
8,985,010 |
|
The accompanying notes are an integral part of these Consolidated Financial Statements.
F-5
Corbus Pharmaceuticals Holdings, Inc.
Consolidated Statements of Cash Flows
|
|
|
Year Ended December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(8,850,739 |
) |
|
$ |
(2,540,485 |
) |
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
|
|
|
Share-based compensation expense |
|
|
1,153,302 |
|
|
|
193,120 |
|
|
Depreciation |
|
|
43,943 |
|
|
|
10,405 |
|
|
Gain on foreign exchange |
|
|
(1,977 |
) |
|
|
(4,570 |
) |
|
Gain on settlement of notes payable, accrued interest and accounts payable |
|
|
— |
|
|
|
(145,006 |
) |
|
Changes in fair value of derivative warrant liability |
|
|
— |
|
|
|
28,448 |
|
|
Forgiveness of interest on notes payable |
|
|
— |
|
|
|
(7,466 |
) |
|
Non-cash interest expense |
|
|
— |
|
|
|
29,861 |
|
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
|
|
|
Increase in restricted cash |
|
|
(22,647 |
) |
|
|
(13,728 |
) |
|
Increase in prepaid expenses |
|
|
(105,959 |
) |
|
|
(268,056 |
) |
|
Increase in accounts payable |
|
|
972,194 |
|
|
|
170,239 |
|
|
Increase in accrued expenses |
|
|
312,788 |
|
|
|
186,002 |
|
|
Increase in deferred revenue |
|
|
1,851,618 |
|
|
|
— |
|
|
Net cash used in operating activities |
|
|
(4,647,477 |
) |
|
|
(2,361,236 |
) |
|
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
Purchases of property and equipment |
|
|
(114,037 |
) |
|
|
(64,448 |
) |
|
Net cash used in investing activities |
|
|
(114,037 |
) |
|
|
(64,448 |
) |
|
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
Proceeds from issuance of notes payable |
|
|
207,750 |
|
|
|
192,000 |
|
|
Principal payments on notes payable |
|
|
(190,120 |
) |
|
|
(247,006 |
) |
|
Proceeds from issuance on common stock |
|
|
11,328,929 |
|
|
|
10,305,161 |
|
|
Issuance costs incurred common stock financings |
|
|
(509,215 |
) |
|
|
(1,865,046 |
) |
|
Net cash provided by financing activities |
|
|
10,837,344 |
|
|
|
8,385,109 |
|
|
Net increase in cash and cash equivalents |
|
|
6,075,830 |
|
|
|
5,959,425 |
|
|
Cash and cash equivalent at beginning of the period |
|
|
6,262,445 |
|
|
|
303,020 |
|
|
Cash and cash equivalent at end of the period |
|
$ |
12,338,275 |
|
|
$ |
6,262,445 |
|
|
Supplemental disclosure of cash flow information and non cash transactions: |
|
|
|
|
|
|
|
|
|
Conversion of Series A preferred stock into common stock |
|
$ |
— |
|
|
$ |
1,108,609 |
|
|
Conversion of notes payable, accrued interest and accounts payable into common stock and a warrant |
|
$ |
— |
|
|
$ |
396,000 |
|
|
Reclassification of derivative warrant liability to equity |
|
$ |
— |
|
|
$ |
48,380 |
|
The accompanying notes are an integral part of these Consolidated Financial Statements.
F-6
Corbus Pharmaceuticals Holdings, Inc.
Notes to Consolidated Financial Statements
|
1. |
NATURE OF OPERATIONS |
Business
Corbus Pharmaceuticals Holdings, Inc. (“CPHI” or “the Company”) is a clinical stage pharmaceutical company, focused on the development and commercialization of novel therapeutics to treat rare, chronic, and serious inflammatory and fibrotic diseases. Since its inception, the Company has devoted substantially all of its efforts to business planning, research and development, recruiting management and technical staff, acquiring operating assets and raising capital. The Company’s business is subject to significant risks and uncertainties and the Company will be dependent on raising substantial additional capital before it becomes profitable and it may never achieve profitability.
Reverse Acquisition
JB Therapeutics Inc. (“JB Therapeutics”), was incorporated on April 24, 2009 under the laws of the State of Delaware. CPHI was incorporated on December 18, 2013 under the laws of the State of Delaware. On April 11, 2014, JB Therapeutics Inc. completed a reverse acquisition with CPHI. Upon the consummation of the reverse acquisition, JB Therapeutics became a wholly owned subsidiary of CPHI and changed its name to Corbus Pharmaceuticals, Inc. CPHI continues to operate the business of JB Therapeutics. All the references to the “Company” for activities prior to the reverse merger are to JB Therapeutics activities.
As part of the reverse acquisition, CPHI issued 9,000,000 shares of common stock to holders of JB Therapeutics common and preferred stock in exchange for a total of 5,964,649 common shares outstanding on an as converted basis. In addition, the holders of warrants to purchase common stock of JB Therapeutics received warrants, or the Replacement Warrants, to purchase 377,839 shares of CPHI common stock with an exercise price of ranging from $0.60 to $0.66. In addition, holders of JB Therapeutics Series A preferred stock received warrants to purchase 917,612 shares of common stock of CPHI. Finally, holders of outstanding options of JB Therapeutics received, in substitution for such options, options made pursuant to the Corbus 2014 Equity Incentive Plan to purchase an aggregate of 905,334 shares of CPHI common stock with exercise prices ranging from $0.11 to $0.17 per share. All share and per share amounts presented in these consolidated financial statements for the year ended December 31, 2014 have been retroactively restated to reflect the 1.5089 exchange ratio of JB Therapeutics shares for CPHI shares in the reverse acquisition. Immediately prior to the reverse acquisition, CPHI had 6,000,000 shares outstanding.
The reverse acquisition was accounted for as a recapitalization since the formation of CPHI was formed solely to effect the reverse acquisition and a private placement of equity, and CPHI had no prior operations or net monetary assets. Thus, JB Therapeutics is deemed to be the accounting acquirer and successor entity and the historical financial statements are those of JB Therapeutics as the accounting acquirer. Following the reverse acquisition, the management of JB Therapeutics became the management of CPHI. At the date of the reverse acquisition, the 6,000,000 outstanding shares of CPHI were reflected as an issuance of common stock to the prior holders of CPHI. CPHI had no net monetary assets as of the reverse acquisition so the issuance was recorded as a reclassification between additional paid-in-capital and par value of common stock. On April 11, 2014 and in three subsequent closings, CPHI completed a private placement of equity, raising net proceeds of approximately $8,402,000 (the “2014 Private Placement”) (See Note 12).
|
2. |
LIQUIDITY AND GOING CONCERN |
The accompanying consolidated financial statements have been prepared assuming the Company will continue as a going concern, which contemplates continuity of operations, realization of assets and the satisfaction of liabilities and commitments in the normal course of business. The Company has incurred recurring losses since inception and as of December 31, 2015, had an accumulated deficit of approximately $13,278,000. The Company anticipates operating losses to continue for the foreseeable future due to, among other things, costs related to research funding, development of its product candidates and its preclinical programs, strategic alliances and the development of its administrative organization. The Company expects the current cash on hand of $12,338,275 to be sufficient to meet its operating and capital requirements into the fourth quarter of 2016 based on planned expenditures. Should the Company be unable to raise sufficient additional capital, the Company may undertake cost-cutting measures including delaying or discontinuing certain clinical activities. The Company will need to raise significant additional capital to fund the clinical trials for Resunab. The Company may seek to sell common or preferred equity or convertible debt securities, enter into a credit facility or another form of third-party funding, or seek other debt financing. The sale of equity and convertible debt securities may result in dilution to the Company’s stockholders and those securities may have rights senior to those of the Company’s common shares. If the Company raises additional funds through the issuance of preferred stock, convertible debt securities or other debt financing, these securities or other debt could contain covenants that would restrict the Company’s operations. Any other third-party funding arrangement could require the Company to relinquish valuable rights.
F-7
The source, timing and availability of any future financing will depend principally upon market conditions, and, more specifically, on the progress of the Company’s clinical development programs. Funding may not be available when needed, at all, or on terms acceptable to the Company. Lack of necessary funds may require the Company, among other things, to delay, scale back or eliminate some or all of the Company’s planned clinical trials. There have been no adjustments made to these consolidated financial statements as a result of these uncertainties.
|
3. |
SIGNIFICANT ACCOUNTING POLICIES |
A summary of the significant accounting policies followed by the Company in the preparation of the financial statements is as follows:
Use of Estimates
The process of preparing financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of assets and liabilities at the date of financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates and changes in estimates may occur. The most significant estimates are related to stock based compensation, the value of derivative instruments and the accrual of research and clinical obligations.
Prior to the registration of its common stock and the subsequent public listing of the common stock, the Company had granted stock options at exercise prices not less than the fair value of its common stock as determined by the board of directors, with input from management. The Company’s board of directors determined the estimated fair value of the common stock based on a number of objective and subjective factors, including external market conditions affecting the biotechnology industry sector and the historic prices at which the Company sold shares of preferred stock.
Cash and Cash Equivalents
The Company considers only those investments which are highly liquid, readily convertible to cash, and that mature within three months from date of purchase to be cash equivalents. Marketable investments are those with original maturities in excess of three months. At December 31, 2015 and 2014, cash equivalents were comprised of money market funds. The Company had no marketable investments at December 31, 2015 and 2014. Cash and cash equivalents consist of the following:
|
|
|
December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
Cash |
|
$ |
255,943 |
|
|
$ |
10,974 |
|
|
Money market fund |
|
|
12,082,332 |
|
|
|
6,251,471 |
|
|
|
|
$ |
12,338,275 |
|
|
$ |
6,262,445 |
|
Restricted Cash
Restricted cash as of December 31, 2015 and December 31, 2014 was $36,375 and $13,728, respectively, and was related to a stand-by letter of credit in favor of a landlord (See Note 6).
Financial Instruments
The carrying amounts reported in the consolidated balance sheet for cash and cash equivalents and accounts payable approximate fair value based on the short-term nature of these instruments. The carrying value of loans payable approximate their fair value due to their market terms.
Property and Equipment
The estimated life for all Property and Equipment is 3 years, except for leasehold improvements which are amortized over the life of the lease. See Note 6 for Operating Lease Commitment.
F-8
Research and Development Expenses and Collaborative Research Agreements
Costs incurred for research and development are expensed as incurred. For periods prior to 2015, the Company recorded payments received from research and development grants and awards as a reduction in research and development expense in the statement of operations.
For the development award received from the Cystic Fibrosis Foundation during 2015 (See Note 17), the Company is recognizing amounts received as revenue under this collaborative research agreement in accordance with the milestone method of Accounting Standards Codification 605—Revenue Recognition, under which payments are recognized as revenue in their entirety when a related milestone is achieved. The research grants prior to 2015 were immaterial, therefore they were not re-classified to revenue.
Accruals for Research and Development Expenses and Clinical Trials
As part of the process of preparing its financial statements, the Company is required to estimate its expenses resulting from its obligations under contracts with vendors, clinical research organizations and consultants and under clinical site agreements in connection with conducting clinical trials. The financial terms of these contracts are subject to negotiations, which vary from contract to contract and may result in payment flows that do not match the periods over which materials or services are provided under such contracts. The Company’s objective is to reflect the appropriate trial expenses in its financial statements by matching those expenses with the period in which services are performed and efforts are expended. The Company accounts for these expenses according to the timing of various aspects of the trial. The Company determines accrual estimates through financial models taking into account discussion with applicable personnel and outside service providers as to the progress or state of consummation of trials, or the services completed. During the course of a clinical trial, the Company adjusts its clinical expense recognition if actual results differ from its estimates. The Company makes estimates of its accrued expenses as of each balance sheet date based on the facts and circumstances known to it at that time. The Company’s clinical trial accruals are dependent upon the timely and accurate reporting of contract research organizations and other third-party vendors. Although the Company does not expect its estimates to be materially different from amounts actually incurred, its understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and may result in it reporting amounts that are too high or too low for any particular period. For the years ended December 31, 2015 and 2014, there were no material adjustments to the Company’s prior period estimates of accrued expenses for clinical trials.
Concentrations of Credit Risk
The Company has no significant off-balance-sheet concentration of credit risk such as foreign exchange contracts, option contracts or other hedging arrangements. The Company may from time to time have cash in banks in excess of Federal Deposit Insurance Corporation insurance limits.
Segment Information
Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision maker, or decision making group, in making decisions regarding resource allocation and assessing performance. To date, the Company has viewed its operations and manages its business as principally one operating segment, which is developing and commercializing therapeutics to treat rare life-threatening inflammatory fibrotic diseases. As of December 31, 2015 and 2014, all of the Company’s assets were located in the United States.
Income Taxes
For federal and state income taxes, deferred tax assets and liabilities are recognized based upon temporary differences between the financial statement and the tax basis of assets and liabilities. Deferred income taxes are based upon prescribed rates and enacted laws applicable to periods in which differences are expected to reverse. A valuation allowance is recorded to reduce a net deferred tax benefit when it is more likely than not that the tax benefit from the deferred tax assets will not be realized. Accordingly, given the cumulative losses since inception, the Company has provided a valuation allowance equal to 100% of the tax benefit in order to eliminate the deferred tax assets amounts. Tax positions taken or expected to be taken in the course of preparing the Company’s tax returns are required to be evaluated to determine whether the tax positions are “more-likely-than-not” of being sustained by the applicable tax authority.
Tax positions not deemed to meet a more-likely-than-not threshold, as well as accrued interest and penalties, if any, would be recorded as a tax expense in the current year. There were no uncertain tax positions that require accrual or disclosure to the financial statements as of December 31, 2015 or 2014.
F-9
Impairment of Long-lived Assets
The Company continually monitors events and changes in circumstances that could indicate that carrying amounts of long-lived assets may not be recoverable. An impairment loss is recognized when expected cash flows are less than an asset’s carrying value. Accordingly, when indicators of impairment are present, the Company evaluates the carrying value of such assets in relation to the operating performance and future undiscounted cash flows of the underlying assets. The Company’s policy is to record an impairment loss when it is determined that the carrying value of the asset may not be recoverable. No impairment charges were recorded for the years ended December 31, 2015 and 2014.
Share-based Payments
The Company recognizes compensation costs resulting from the issuance of stock-based awards to employees, non-employees and directors as an expense in the statement of operations over the service period based on a measurement of fair value for each stock-based award. The fair value of each option grant is estimated as of the date of grant using the Black-Scholes option-pricing model. The fair value is amortized as compensation cost on a straight-line basis over the requisite service period of the awards, which is generally the vesting period. Stock options granted to non-employee consultants are revalued at the end of each reporting period until vested and the changes in their fair value are recorded as adjustments to expense over the related vesting period.
Derivative Instruments
The Company generally does not use derivative instruments to hedge exposures to cash-flow or market risks; however, certain warrants to purchase common stock that do not meet the requirements for classification as equity are classified as liabilities. In such instances, net-cash settlement is assumed for financial reporting purposes, even when the terms of the underlying contracts do not provide for a net-cash settlement. Such financial instruments are initially recorded at fair value with subsequent changes in fair value charged (credited) to operations in each reporting period. If these instruments subsequently meet the requirements for classification as equity, the Company reclassifies the fair value to equity.
Net Loss Per Common Share
Basic net loss per share of the Company’s common stock has been computed by dividing net loss by the weighted average number of shares outstanding during the period. Diluted net income per share of the Company’s common stock has been computed by dividing net income by the weighted average number of shares outstanding plus the dilutive effect, if any, of outstanding stock options, warrants and convertible securities. Diluted net loss per share of the Company’s common stock has been computed by dividing the net loss for the period by the weighted average number of shares of the Company’s common stock outstanding during such period. For years in which there is a net loss, options, warrants and convertible securities are anti-dilutive and therefore excluded from diluted loss per share calculations. The following table sets forth the computation of basic and diluted earnings per share for the years ended December 31, 2015 and 2014:
|
|
|
Years Ended December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
Basic and diluted net loss per share of common stock: |
|
|
|
|
|
|
|
|
|
Net loss |
|
$ |
(8,850,739 |
) |
|
$ |
(2,540,485 |
) |
|
Net loss applicable to common stockholders |
|
|
(8,850,739 |
) |
|
|
(2,540,485 |
) |
|
Weighted average shares of common stock outstanding |
|
|
31,350,145 |
|
|
|
20,159,861 |
|
|
Net loss per share of common stock-basic and diluted |
|
$ |
(0.28 |
) |
|
$ |
(0.13 |
) |
The following potentially dilutive securities outstanding at December 31, 2015 and 2014 have been excluded from the computation of dilutive weighted average shares outstanding as the inclusion would be antidilutive.
|
|
|
December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
Warrants |
|
|
1,969,250 |
|
|
|
13,709,977 |
|
|
Stock options |
|
|
3,982,065 |
|
|
|
3,556,691 |
|
|
|
|
|
5,951,315 |
|
|
|
17,266,668 |
|
F-10
Recent Accounting Pronouncements
Accounting for Share-Based Payments
In June 2014, the Financial Accounting Standards Board (the “FASB”) issued Accounting Standards Update (“ASU”) No. 2014-12, Accounting for Share-Based Payments When the Terms of an Award Provide That a Performance Target Could Be Achieved after the Requisite Service Period (a consensus of the FASB Emerging Issues Task Force) ("ASU 2014-12"). ASU 2014-12 clarifies that entities should treat performance targets that can be met after the requisite service period of a share-based payment award as performance conditions that affect vesting. Therefore, an entity would not record compensation expense (measured as of the grant date without taking into account the effect of the performance target) related to an award for which transfer to the employee is contingent on the entity's satisfaction of a performance target until it becomes probable that the performance target will be met. There are no new disclosures required under ASU 2014-12. ASU 2014-12 is effective for fiscal years, and interim periods within those years, beginning after December 15, 2015. The Company believes that the adoption of ASU 2014-12 will not have a material impact on its financial position, results of operations, cash flows, or disclosures.
Reporting of Going-Concern Uncertainties
In August 2014, the FASB issued ASU No. 2014-15, Presentation of Financial Statements—Going Concern (“ASU 2014-15), which states management should evaluate whether there are conditions or events, considered in the aggregate, that raise a substantial doubt about the entity’s ability to continue as a going concern within one year after the date that the financial statements are issued. Management’s evaluation should be based on relevant conditions and events that are known and likely to occur at the date that the financial statements are issued. ASU 2014-15 will be effective for the annual period ending after December 15, 2016, and for annual periods and interim periods thereafter, however, early application is permitted. Management does not expect the adoption of ASU 2014-15 to have material impact on the Company’s consolidated financial statements, although there may be additional disclosures upon adoption.
Accounting for Leases
In February 2016, the FASB issued ASU 2016-02, Leases (Topic 842) (“ASU 2016-02”). Under ASU 2016-02, a lessee will be required to recognize assets and liabilities for leases with lease terms of more than 12 months. Consistent with current GAAP, the recognition, measurement, and presentation of expenses and cash flows arising from a lease by a lessee primarily will depend on its classification as a finance or operating lease. However, unlike current GAAP, which requires only capital leases to be recognized on the balance sheet, ASU 2016-02 will require both types of leases to be recognized on the balance sheet. ASU 2016-02 will take effect for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2018, with early application permitted. Management does not expect the adoption of ASU 2016-02 to have material impact on the Company’s consolidated financial statements, although there may be additional disclosures upon adoption.
|
4. |
FAIR VALUE OF ASSETS AND LIABILITIES |
The Company groups its assets and liabilities measured at fair value in three levels, based on the markets in which the assets and liabilities are traded and the reliability of the assumptions used to determine fair value.
Level 1—Valuation is based on quoted prices in active markets for identical assets or liabilities. Level 1 assets and liabilities generally include debt and equity securities that are traded in an active exchange market. Valuations are obtained from readily available pricing sources for market transactions involving identical assets or liabilities.
Level 2—Valuation is based on observable inputs other than Level 1 prices, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities.
Level 3—Valuation is based on unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. Level 3 assets and liabilities include financial instruments whose value is determined using pricing models, discounted cash flow methodologies, or similar techniques, as well as instruments for which the determination of fair value requires significant management judgment or estimation.
The Company uses valuation methods and assumptions that consider, among other factors, the fair value of the underlying stock, risk-free interest rate, volatility, expected life and dividend rates in estimating fair value for the warrants considered to be derivative instruments. The Company had no assets or liabilities classified as Level 1 or Level 2. Certain warrants issued for professional services (Note 14) were classified as Level 3. The fair values of these instruments were determined using models based on market observable inputs and management judgment. There were no material re-measurements of fair value during the years ended December 31, 2015 and 2014 with respect to financial assets and liabilities, other than those assets and liabilities that are measured at fair value on a recurring basis.
F-11
The Company had valued certain warrants as a derivative liability at December 31, 2013 and due to a modification in the terms of these warrants (See Note 14), the derivative liability was reclassified at June 30, 2014 to Additional Paid in Capital. The warrant derivative liability was re-measured at June 30, 2014 prior to reclassification using the Black-Scholes option pricing model based on the following assumptions:
|
|
|
As of June 30, 2014 |
|
|
|
Risk free interest rate |
|
|
1.25 |
% |
|
Expected dividend yield |
|
|
0 |
% |
|
Contractual term |
|
|
3.97 |
|
|
Expected volatility |
|
|
66 |
% |
As of December 31, 2015 and 2014 there were no derivative warrant liabilities.
Assets and liabilities measured at fair value on a recurring basis are summarized below:
|
|
|
December 31, 2014 |
|
|||||||||||||
|
|
|
Level 1 |
|
|
Level 2 |
|
|
Level 3 |
|
|
Total |
|
||||
|
Liabilities: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Derivative warrant liability at December 31, 2013 |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
19,932 |
|
|
$ |
19,932 |
|
|
Change in fair value of the derivative warrant liability |
|
|
— |
|
|
|
— |
|
|
|
28,448 |
|
|
|
28,448 |
|
|
Reclassification of derivative warrant liability to equity |
|
|
— |
|
|
|
— |
|
|
|
(48,380 |
) |
|
|
(48,380 |
) |
|
Derivative warrant liability at December 31, 2014 |
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
$ |
— |
|
|
5. |
PROPERTY AND EQUIPMENT |
Property and Equipment consists of the following:
|
|
|
December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
Computer hardware and software |
|
$ |
40,202 |
|
|
$ |
17,179 |
|
|
Office furniture and equipment |
|
|
35,209 |
|
|
|
27,960 |
|
|
Leasehold improvements |
|
|
19,310 |
|
|
|
19,310 |
|
|
Construction in progress |
|
|
83,765 |
|
|
|
— |
|
|
Less: accumulated depreciation |
|
|
(54,348 |
) |
|
|
(10,405 |
) |
|
Property and equipment, net |
|
$ |
124,138 |
|
|
$ |
54,044 |
|
Depreciation expense was approximately $44,000 and $10,000 for the years ended December 31, 2015 and 2014, respectively. At December 31, 2015, construction in progress consists of purchased property and equipment not placed in service until the Company’s occupation of its new office space in January 2016 (See Note 6).
F-12
|
6. |
COMMITMENTS AND CONTINGENCIES |
Operating Lease Commitment
On May 30, 2014, the Company entered into a commercial lease for 2,387 square feet of office space in Norwood, MA. The lease commenced on July 1, 2014, had a three-year term, and required a standby letter of credit of $13,728 payable in favor of the landlord. In August 2015, the lease was amended for the relocation of the Company into 6,326 square feet of office space within the existing building. In January 2016, the Company began occupying the space under this lease amendment, which is for a five-year term. The amendment also required an increase in the standby letter of credit to $36,375 (See Note 3). Pursuant to the terms of the Company’s non-cancelable lease agreements in effect at December 31, 2015, the future minimum rent commitments are as follows:
|
2016 |
|
$ |
250,768 |
|
|
2017 |
|
|
148,397 |
|
|
2018 |
|
|
151,561 |
|
|
2019 |
|
|
154,723 |
|
|
2020 |
|
|
157,887 |
|
|
Thereafter |
|
|
13,179 |
|
|
Total |
|
$ |
876,515 |
|
Total rent expense for the years ended December 31, 2015 and 2014, including month-to-month leases, was $55,496 and $35,550, respectively.
|
7. |
NOTES PAYABLE |
The Company entered into notes payable agreements with vendors in lieu of making payments due on accounts payable to these vendors. Interest accrued on these interest bearing notes payable at an annual rate of 7% with accrued interest and principal due at maturity. In August 2014, the Company entered into a settlement agreement with a vendor for the repayment of $631,000 which included $531,000 in notes payable, $93,000 of accrued interest and $7,000 of accounts payable. Under the terms of the settlement agreement, the Company paid the vendor $90,000 and issued 541,948 shares of common stock and a warrant to purchase 162,539 shares of common stock exercisable at $1.00 per share with a five year term. The Company valued the common stock at $341,000 and estimated the fair value of the warrant to be $55,000 based on a Black-Sholes valuation. The Company estimated the fair value of the common stock to be $0.63 per share based upon the 2014 Private Placement in which the Company sold units consisting of one share of common stock and one warrant for $1.00 each. The Company estimated the fair value of each warrant to be $0.37 based on a Black Scholes valuation model. For the year ended December 31, 2014, the Company recorded a gain on the settlement of $145,006 which was recorded as other income in the statement of operations.
The Company also had a note payable outstanding to another vendor with a balance due of $75,244 at September 30, 2014 which had no stated interest rate. The Company had been accruing interest on this note but reached an agreement with the vendor to pay off this note payable with no interest in four equal monthly principal installments of $25,081 and the note was paid off as of December 31, 2014.
In October 2014, the Company entered into a loan agreement with a financing company for $192,000. The terms of the loan stipulated equal monthly payments of principal and interest payments of $24,293 over an eight month period. Interest accrued on this loan at an annual rate of 3.25%. The loan was fully repaid in June 2015.
In November 2015, the Company entered into a loan agreement with a financing company for $207,750. The terms of the loan stipulate equal monthly payments of principal and interest payments of $23,397 over a nine month period. Interest accrues on this loan at an annual rate of 3.25%.
Interest expense for notes payable for the years ended December 31, 2015 and 2014 totaled $2,440 and $24,021, respectively.
Notes payable consisted of the following:
|
|
|
December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
Notes payable |
|
$ |
162,019 |
|
|
$ |
144,389 |
|
|
Less: current portion |
|
|
(162,019 |
) |
|
|
(144,389 |
) |
|
|
|
$ |
— |
|
|
$ |
— |
|
F-13
|
8. |
ACCRUED EXPENSES |
Accrued expenses consisted of the following:
|
|
|
December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
Accrued clinical operations and trials costs |
|
$ |
365,188 |
|
|
$ |
— |
|
|
Accrued product development costs |
|
|
152,018 |
|
|
|
114,757 |
|
|
Accrued audit fees |
|
|
16,500 |
|
|
|
50,000 |
|
|
Accrued payroll |
|
— |
|
|
|
40,734 |
|
|
|
Accrued other |
|
|
28,573 |
|
|
|
44,000 |
|
|
Total |
|
$ |
562,279 |
|
|
$ |
249,491 |
|
|
9. |
DEFERRED REVENUE |
In May 2015, the Company received $1,250,000 upon signing the CFFT award agreement and in the fourth quarter of 2015, the Company received $1,250,000 from CFFT upon the achievement of a milestone for dosing the first patient (See Note 3 and Note 17). The Company recorded these amounts as deferred revenue and is amortizing the deferred revenue and recognizing revenue on a straight-line basis over the performance period of the development program, which is expected to conclude during the first quarter of 2017. The Company recorded $648,382 of revenue during the year ended December 31, 2015. Deferred revenue consists of the following:
|
|
|
December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
Deferred revenue |
|
$ |
1,851,618 |
|
|
$ |
— |
|
|
Less: current portion |
|
|
(1,591,358 |
) |
|
|
— |
|
|
Long term portion |
|
|
260,260 |
|
|
|
— |
|
|
10. |
INCOME TAXES |
No provision or benefit for federal or state income taxes has been recorded, as the Company has incurred a net loss for all of the periods presented, and the Company has provided a full valuation allowance against its deferred tax assets.
At December 31, 2015, and 2014, the Company had federal and Massachusetts net operating loss carryforwards of approximately $22,416,000 and $7,164,000, respectively, of which federal carryforwards will expire in varying amounts beginning in 2029. Massachusetts net operating losses began to expire in 2014. Utilization of net operating losses may be subject to substantial annual limitations due to the “change in ownership” provisions of the Internal Revenue Code, and similar state provisions. The annual limitations may result in the expiration of net operating losses before utilization. The Company also had research and development tax credit carryforwards at December 31, 2015 and 2014 of approximately $441,000 and $190,000, respectively.
Significant components of the Company’s net deferred tax asset are as follows:
|
|
|
December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
NOL carryforward |
|
$ |
4,505,965 |
|
|
$ |
1,408,745 |
|
|
Tax credits |
|
|
406,888 |
|
|
|
167,127 |
|
|
Stock based compensation |
|
|
453,906 |
|
|
|
60,373 |
|
|
Other temporary differences |
|
|
12,581 |
|
|
|
128,093 |
|
|
Subtotal |
|
|
5,379,340 |
|
|
|
1,764,338 |
|
|
Valuation allowance |
|
|
(5,379,340 |
) |
|
|
(1,764,338 |
) |
|
Net deferred tax asset |
|
$ |
— |
|
|
$ |
— |
|
F-14
The Company has maintained a full valuation allowance against its deferred tax assets in all periods presented. A valuation allowance is required to be recorded when it is more likely than not that some portion or all of the net deferred tax assets will not be realized. Since the Company cannot determine that it is more likely than not that it will generate taxable income, and thereby realize the net deferred tax assets, a full valuation allowance has been provided. The Company has no uncertain tax positions at December 31, 2015 and 2014 that would affect its effective tax rate. The Company does not anticipate a significant change in the amount of uncertain tax positions over the next twelve months. Since the Company is in a loss carryforward position, the Company is generally subject to U.S. federal and state income tax examinations by tax authorities for all years for which a loss carryforward is available.
Income tax benefits computed using the federal statutory income tax rate differs from the Company’s effective tax rate primarily due to the following:
|
|
|
December 31, |
|
|||||
|
|
|
2015 |
|
|
2014 |
|
||
|
Tax provision at statutory rate |
|
|
34.00 |
% |
|
|
34.00 |
% |
|
State taxes, net of federal benefit |
|
|
4.76 |
% |
|
|
5.03 |
% |
|
Permanent differences |
|
|
-0.62 |
% |
|
|
-0.47 |
% |
|
Tax credits |
|
|
2.67 |
% |
|
|
3.37 |
% |
|
Other |
|
|
0.04 |
% |
|
|
0.21 |
% |
|
Decrease in valuation reserve |
|
|
-40.85 |
% |
|
|
-42.14 |
% |
|
Total |
|
|
0.00 |
% |
|
|
0.00 |
% |
|
11. |
SERIES A PREFERRED STOCK |
In April 2014, in conjunction with the reverse acquisition and financing, 1,835,212 shares of JB Therapeutics’ Series A Convertible Preferred Stock and 200,000 shares of its Series A Non-Convertible Preferred Stock were converted into 2,035,212 shares of the Company’s common stock.
Prior to the conversion, the Company had classified the Series A Convertible and Series A Non-convertible Preferred Stock outside of permanent equity based upon the terms of the instruments.
|
12. |
COMMON STOCK |
The Company has authorized 150,000,000 shares of common stock, $0.0001 par value per share, of which 37,605,134 shares and 25,938,332 shares were issued and outstanding as of December 31, 2015 and 2014, respectively.
On April 11, 2014, in conjunction with the reverse acquisition (See Note 1) and the 2014 Private Placement, the Company issued 6,000,000 shares of common stock to the former shareholders of CPHI and 1,835,212 shares of Series A Convertible Preferred Stock and 200,000 shares of Series A Non-Convertible Preferred Stock were converted into 2,035,212 shares of common stock.
In August 2014, the Company issued 541,948 shares of common stock to a vendor in connection with a settlement of debt obligation (Note 7).
In December 2014, the Company issued 87,691 shares of common stock upon the exercise of stock options to purchase common stock and the Company received net proceeds of $4,450.
In December 2014, the Company issued 33,333 shares of common stock upon the exercise of warrants to purchase 33,333 shares of common stock and the Company received net proceeds of $33,333. Also in December 2014, the Company issued 15,360 shares of common stock upon the exercise of warrants to purchase 21,500 shares of common stock that were exercised under a cashless exercise provision.
During the year ended December 31, 2015, the Company issued 11,666,802 shares of common stock upon the exercise of stock options and warrants to purchase common stock and the Company received net proceeds of $10,819,714 from these exercises.
2014 Private Placement
In four closings in April and May 2014, CPHI completed the 2014 Private Placement and sold an aggregate of 10,260,000 shares of common stock and warrants to purchase an aggregate of 10,260,000 shares of CPHI common stock with an exercise price of $1.00 per share and a five year term. The aggregate gross proceeds of $10,260,000 were allocated between the common stock and the warrants based on their relative fair values which amounted to $5,349,000 for the common stock and $4,911,000 for the warrants.
F-15
Net proceeds after deducting offering expenses were approximately $8,402,000. Aegis Capital Corp acted as the exclusive placement agent for the 2014 Private Placement (the “Placement Agent”). The Placement Agent received a cash fee of $1,023,000 and a non-accountable expense allowance of $308,000. In addition, the Placement Agent received warrants to purchase 2,047,000 shares of common stock with an exercise price of $1.00 per share and a five year term. These warrants contain a cashless exercise feature and the fair value of the warrants was recorded as a stock issuance cost and was netted against the gross proceeds received. Certain members of the management of Aegis Capital Corp. are also shareholders of CPHI.
In connection with the 2014 Private Placement, CPHI entered into a registration rights agreement with the private placement investors, the Placement Agent and holders of its outstanding warrants. CPHI was required to file a registration statement with the Securities and Exchange Commission (“SEC”) no later than July 29, 2014, covering the resale of the shares of common stock and the shares of common stock underlying the warrants issued in the 2014 Private Placement, as well as the existing warrants. The Company filed a confidential registration statement on Form S-1 on July 2, 2014 and the S-1 was declared effective by the SEC on October 3, 2014. The Company was required to keep the registration statement effective for a period of one year, or until Rule 144 of the Securities Act became available to investors, whichever was earlier.
|
13. |
STOCK OPTIONS |
In April 2014, the Company adopted the Corbus Pharmaceticals Holdings, Inc. 2014 Equity Incentive Plan (the “2014 Plan”). Pursuant to the 2014 Plan, the Company’s Board of Directors may grant incentive and nonqualified stock options and restricted stock to employees, officers, directors, consultants and advisors. On January 1, 2015, pursuant to an annual evergreen provision contained in the 2014 Plan, the number of shares reserved for future grants was increased by 1,815,683 shares. As of December 31, 2015, there was a total of 8,666,017 shares reserved for issuance under the 2014 plan and there were 4,545,135 shares available for future grants. Options issued under the 2014 Plan are exercisable for up to 10 years from the date of issuance.
Pursuant to the terms of an annual evergreen provision in the 2014 Plan, the number of shares of common stock available for issuance under the 2014 Plan shall automatically increase on January 1 of each year by at least seven percent (7%) of the total number of shares of common stock outstanding on December 31st of the preceding calendar year, or, pursuant to the terms of the 2014 Plan, in any year, the Board of Directors may determine that such increase will provide for a lesser number of shares. In accordance with the terms of the 2014 Plan, effective as of January 1, 2016, the Board of Directors approved an increase in the number of shares of common stock available for issuance under the 2014 Plan in an amount of 1,250,000 shares, such amount being less than seven percent (7%) of the outstanding shares of common stock on December 31, 2015. As of January 1, 2016, the 2014 Plan had a total reserve of 9,916,017 shares and there were 5,795,133 shares available for future grants.
Share-based Compensation
For stock options issued and outstanding for the years ended December 31, 2015 and 2014, the Company recorded non-cash, stock-based compensation expense of $1,153,302 and $193,120, respectively, net of estimated forfeitures.
The fair value of each option award is estimated on the date of grant using the Black-Scholes option pricing model that uses the assumptions noted in the following table. Due to its limited operating history and limited number of sales of its common stock, the Company estimated its volatility in consideration of a number of factors, including the volatility of comparable public companies and, commencing in 2015, the Company also included the volatility of its own common stock. The Company uses historical data, as well as subsequent events occurring prior to the issuance of the financial statements, to estimate option exercises and employee terminations within the valuation model. The expected term of options granted under the 2014 Plan, all of which qualify as “plain vanilla” per SEC Staff Accounting Bulletin 107, is based on the average of the 6.25 years. For non-employee options, the expected term is the contractual term. The risk-free rate is based on the yield of a U.S. Treasury security with a term consistent with the option.
The weighted average assumptions used principally in determining the fair value of options granted were as follows:
|
|
|
2015 |
|
|
2014 |
|
||
|
Risk free interest rate |
|
|
1.85 |
% |
|
|
1.88 |
% |
|
Expected dividend yield |
|
|
0 |
% |
|
|
0 |
% |
|
Expected term in years |
|
|
6.73 |
|
|
|
6.20 |
|
|
Expected volatility |
|
|
90.68 |
% |
|
|
89.75 |
% |
|
Estimated forfeiture rate |
|
|
4.83 |
% |
|
|
19.20 |
% |
F-16
A summary of option activity for years ended December 31, 2015 and 2014 is presented below:
|
Options |
|
Shares |
|
|
Weighted Average Exercise Price |
|
|
Weighted Average Remaining Contractual Term in Years |
|
|
Average Intrinsic Value |
|
||||
|
Outstanding at December 31, 2013 |
|
|
597,243 |
|
|
$ |
0.13 |
|
|
|
|
|
|
|
|
|
|
Granted |
|
|
3,047,139 |
|
|
$ |
0.95 |
|
|
|
|
|
|
|
|
|
|
Exercised |
|
|
(87,691 |
) |
|
$ |
0.11 |
|
|
|
|
|
|
|
|
|
|
Outstanding at December 31, 2014 |
|
|
3,556,691 |
|
|
$ |
0.83 |
|
|
|
|
|
|
|
|
|
|
Granted |
|
|
600,002 |
|
|
$ |
2.10 |
|
|
|
|
|
|
|
|
|
|
Exercised |
|
|
(51,128 |
) |
|
$ |
0.11 |
|
|
|
|
|
|
$ |
152,531 |
|
|
Forfeited |
|
|
(123,500 |
) |
|
$ |
1.00 |
|
|
|
|
|
|
|
|
|
|
Outstanding at December 31, 2015 |
|
|
3,982,065 |
|
|
$ |
1.03 |
|
|
|
8.10 |
|
|
$ |
2,578,843 |
|
|
Vested at December 31, 2015 |
|
|
1,946,810 |
|
|
$ |
0.87 |
|
|
|
7.63 |
|
|
$ |
1,560,372 |
|
The weighted average grant-date fair value of options granted during the year ended December 31, 2015 was $1.41 per share. The total fair value of options that were vested as of December 31, 2015 was $1,290,500. As of December 31, 2015 there was approximately $1,306,093 of total unrecognized compensation expense, related to non-vested share-based option compensation arrangements. The unrecognized compensation expense is estimated to be recognized over a period of 3.21 years at December 31, 2015.
|
14. |
DERIVATIVE INSTRUMENTS |
In 2013, the Company’s predecessor, JB Therapeutics, Inc., issued warrants for the purchase of 301,778 shares of common stock, which had provisions that included anti-dilution protection, cashless exercise provisions and, under certain conditions, granted holders the right to request the Company repurchase the warrant. Accordingly, these warrants were considered derivative instruments through June 30, 2014 and were recorded as a derivative warrant liability. On June 30, 2014, these warrant agreements were modified to eliminate the anti-dilution protection and accordingly these warrants were no longer considered a derivative liability and the fair value of $48,380 shares was reclassified and recorded as an increase in additional paid-in capital.
To value the derivative warrant liability, the Company used the Black-Scholes option pricing model and assumptions that considered, among other factors, the fair value of the underlying stock, risk-free interest rate, volatility, expected life and dividend rates in estimating fair value for the warrants considered to be derivative instruments. Changes in fair value of the derivative financial instruments were recognized in the statement of operations as gain or loss on change in fair value of warrant liability. There was a loss on change in fair value of the warrant liability of $28,448 recognized for the year ended December 31, 2014.
|
15. |
WARRANTS |
In June 2013, the Company issued warrants to purchase 301,778 shares of common stock at an exercise price of $0.66 per share in exchange for services provided. These warrants were valued at $21,910 in June 2013 and expensed to general and administrative expenses with an offset to derivative liability. On June 30, 2014, the Company issued an additional 48,222 warrants to the warrant holder in exchange for a settlement of a claim and modification of the warrant terms and to eliminate the anti-dilution protection (See Note 14).
In connection with the 2014 Private Placement (See Note 12) the Company issued warrants to investors and former holders of Series A Convertible Preferred Stock to purchase an aggregate of 11,177,612 shares of common stock with an exercise price of $1.00 per share and a five year term. These warrants had a provision that permitted the Company to call and redeem the warrants after a thirty day notice period if the stock traded at $2.50 or greater on a volume weighted average basis for at least twenty consecutive trading days. In addition, the Company issued warrants to the Placement Agent to purchase 2,047,000 shares of common stock with an exercise price of $1.00 per share and a five year term. These warrants contain a cashless exercise feature and the fair value of the warrants was recorded as a stock issuance cost and netted against the gross proceeds received.
In August 2014, the Company issued a warrant to a vendor in connection with a debt settlement (See Note 7) for the purchase of 162,539 shares of common stock exercisable at $1.00 per share with a five year term. The Company estimated the fair value of the warrant to be $55,000 based on a Black-Sholes valuation and charged additional paid-in capital.
F-17
At December 31, 2015, there were warrants outstanding to purchase 1,969,250 shares of common stock with a weighted average exercise price of $0.97 and a weighted average remaining life of 3.33 years. During the year ended December 31, 2015, warrants to purchase 11,615,674 shares of common stock were exercised for net proceeds (see Note 12) of approximately $10,814,125 which included warrants to purchase 371,250 shares of common stock that were exercised in cashless exercises, in accordance with the warrant agreements, resulting in the issuance of 255,724 shares. There were no warrants issued or cancelled during the year ended December 31, 2015.
|
16. |
RELATED PARTY TRANSACTIONS |
In connection with the formation of Corbus Pharmaceutical Holdings, Inc. in December 2013, certain affiliates of Aegis Capital Corp. (the “Placement Agent”) and certain other parties not affiliated with us or the Placement Agent subscribed for an aggregate of 6,000,000 shares of common stock for which they paid an aggregate of $120,000 ($0.02 per share), including David Hochman, one of our directors who purchased 450,000 shares and whose family trust purchased 90,000 shares of common stock.
Following the Initial Closing of the 2014 Private Placement, which took place on April 11, 2014, the Placement Agent had a right to appoint one member of the Company’s board of directors for a two-year term (the “Aegis Nominee”). David Hochman was appointed as the Aegis Nominee.
On June 21, 2014, the Company entered into a consulting agreement with Orchestra Medical Ventures, LLC (“Orchestra”), of which David Hochman is Managing Partner. The agreement provided that Orchestra would render a variety of consulting and advisory services relating principally to identifying and evaluating strategic relationships, licensing opportunities, and business strategies. Orchestra was compensated at a rate of $5,000 per month for twelve months, payable quarterly in advance. During the years ended December 31, 2015 and 2014, the Company paid Orchestra $15,000 and $45,000, respectively. The consulting agreement expired on April 11, 2015 and the Company is not obligated to make future payments.
The Company entered into a non-exclusive financial advisory agreement with Aegis under which the Company paid Aegis $200,000 upon the execution of the agreement, which commenced on September 1, 2015 and expired on November 30, 2015. The Company also paid Aegis a warrant solicitation fee of $309,215 in connection with the exercise of warrants that were called and exercised in the third quarter of 2015.
One of the members of the Company’s scientific advisory board was considered an affiliate of the Company as he owned more than 10% of the Company’s common stock as of December 31, 2015.
|
17. |
DEVELOPMENT AWARD |
On April 20, 2015, the Company entered into an award agreement with the CFFT, a non-profit drug discovery and development affiliate of the Cystic Fibrosis Foundation, pursuant to which it received a development award (the “Award”) for up to $5 million in funding. The funding from the Award is supporting a first-in-patient Phase 2 clinical trial of the Company’s oral anti-inflammatory drug Resunab in adults with cystic fibrosis (“CF”). Upon the execution of the Award agreement, the Company received a payment of $1,250,000 in May 2015 from the CFFT. In the fourth quarter of 2015, the Company received a second payment of $1,250,000 from the CFFT upon the achievement of a milestone for dosing the first patient (See Notes 3 and 9). In 2015, the Company recorded these amounts received from the CFFT totaling $2,500,000 as deferred revenue. The Company is amortizing these amounts on a straight-line basis over the expected duration of the performance period of the development program under the award, which is expected to conclude in the first quarter of 2017. The remaining $2,500,000 under the Award will be paid to the Company incrementally upon the achievement of certain milestones related to the progress of the Phase 2 CF clinical trial, as set forth in the Award agreement.
Pursuant to the terms of the Award agreement, the Company is obligated to make royalty payments to CFFT contingent upon commercialization of Resunab in the Field of Use (as defined in the Award agreement) including a royalty payment equal to five times the amount the Company receives under the Award agreement, up to $25 million, payable in three equal annual installments following the first commercial sale of Resunab, the first of which is due within 90 days following the first commercial sale of Resunab. The Company is also obligated to make a royalty payment to CFFT equal to the amount the Company receives under the Award agreement, up to $5 million, due in the first calendar year in which the aggregate cumulative net sales of Resunab in the Field of Use exceed $500 million. Lastly, the Company is obligated to make royalty payment(s) to CFFT of up to approximately $15 million if the Company transfers, sells or licenses Resunab in the Field of Use other than for certain clinical or development purposes, or if the Company enters into a change of control transaction, with such payment(s) to be credited against the royalty payments due upon commercialization. The Field of Use is defined in the Award as the treatment in humans of CF, asbestosis, bronchiectasis, byssinosis, chronic bronchitis/COPD hypersensitivity pneumonitis, pneumoconiosis, primary ciliary dyskinesis, sarcoidosis and silicosis. Either CFFT or the Company may terminate the agreement for cause, which includes the Company’s material failure to achieve certain commercialization and development milestones. The Company’s payment obligations survive the termination of the Award agreement.
F-18
|
18. |
SUBSEQUENT EVENTS |
Evergreen Provision
Pursuant to the terms of an annual evergreen provision in the 2014 Plan, the number of shares of common stock available for issuance under the 2014 Plan shall automatically increase on January 1 of each year by at least seven percent (7%) of the total number of shares of common stock outstanding on December 31st of the preceding calendar year, or, pursuant to the terms of the 2014 Plan, in any year, the Board of Directors may determine that such increase will provide for a lesser number of shares. In accordance with the terms of the 2014 Plan, effective as of January 1, 2016, the Board of Directors approved an increase in the number of shares of common stock available for issuance under the 2014 Plan in an amount of 1,250,000 shares, such amount being less than seven percent (7%) of the outstanding shares of common stock on December 31, 2015. As of January 1, 2016, the 2014 Plan had a total reserve of 9,916,017 shares and there were 5,795,133 shares available for future grants.
Lupus Development Award
On March 7, 2016, the Company announced that Resunab will be tested for efficacy and safety in a Phase 2 clinical study in systemic lupus erythematosus (“SLE”). The SLE trial has been selected for funding by the National Institutes of Health Autoimmunity Centers of Excellence (ACE) program, through a grant to the Feinstein Institute for Medical Research (FIMR), Manhasset, NY.
The Phase 2 trial will test the efficacy, safety, tolerability and biologic effects of Resunab as a novel, non-immunosuppressive oral treatment to improve signs and symptoms of SLE. The study plans to enroll 100 adult SLE patients with active musculoskeletal disease and will be carried out at approximately ten sites in the United States. These patients will receive either placebo or three different doses of Resunab daily for 84 days with 28 days follow-up.
F-19